

15 October, 2020 by Mojtaba Chardoli. Peer reviewed by Shahriar Lahouti. Last update 15 February 2023.

CONTENTS
- Preface
- Cerebral physiology
- Primary and secondary brain injury
- Definition
- Pathophysiology of secondary brain injury
- Treatment of secondary brain injury
- Media
- Going further
- References
Preface
Acute neurologic disease is bound to get worse. Since the neuronal function is the essence of human existence, preventing the loss of any neural element is a major goal of perioperative anesthesia and critical care management. Traditionally the pathophysiology of brain injuries occurring due to neuroemergencies, such as stroke or head trauma, have been broadly classified into primary brain injury and secondary brain injury. Brain damage that occurs at the onset or immediately after injury (primary brain damage) is simple (in terms of pathology) brain damage caused by a disease or an injury.
However secondary brain injury is not a simple condition, rather its a general term for a variety of pathologies wherein the ongoing brain tissue damage is caused by later reaction of the brain either to the primary injury or to other extracranial causes 1. While secondary brain injury was described in the context of head trauma in the past, this concept is now further expanded to involve other nontraumatic systemic critical conditions wherein the same molecular cascade events can result in brain damage.
As “Time Is The Brain”, It is of paramount importance that front-line providers promptly identify patients with neurologic emergencies and implement appropriate therapeutic and preventive measures to ameliorate the impact of secondary insults to the brain tissue.
In the following discussion the pathophysiology of secondary brain injury and its clinical implication for preventive and therapeutic measures will be covered.
Cerebral physiology
- More than two centuries ago, the Monro-Kellie doctrine characterized the cranial vault as a fixed space comprising 3 components: blood, cerebrospinal fluid (CSF), and brain tissue (figure 1).2
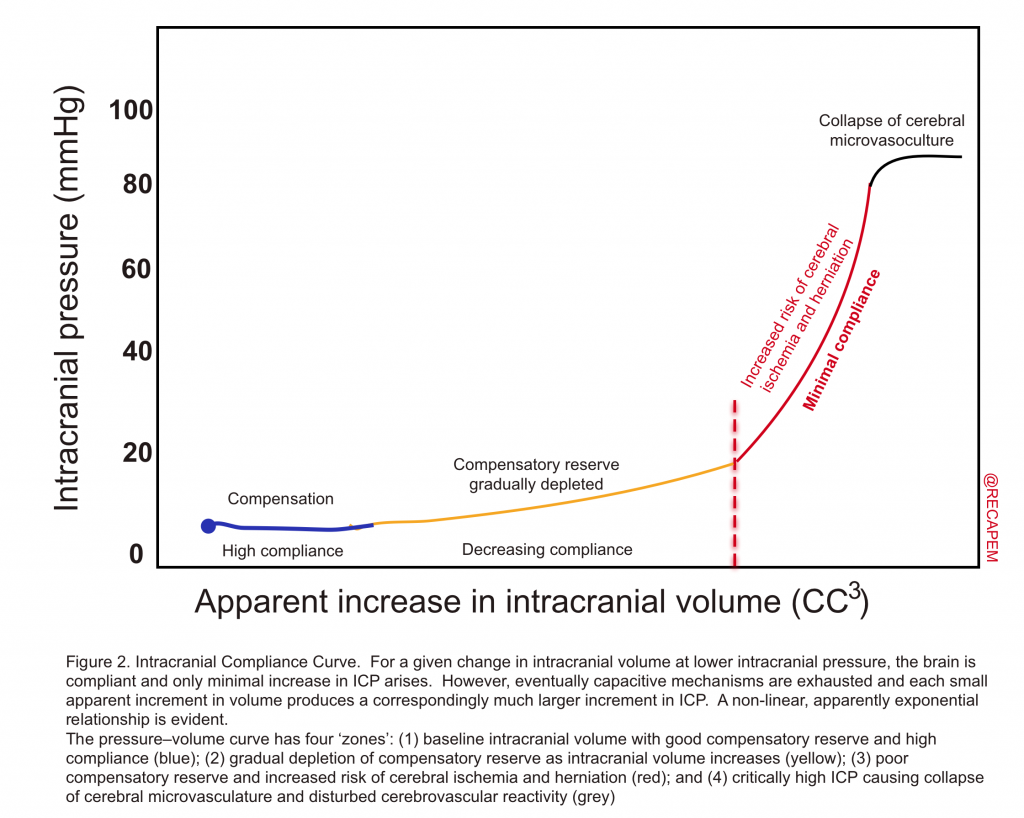
- They defined that the sum of volumes of brain, CSF, and intracranial blood is constant and therefore, an increase in volume in one compartment results in a compensatory decrease in the volume of another. If compensatory mechanisms become exhausted, the intracranial pressure (ICP) increases (figure 2).
- Increase in ICP above a critical level is not tolerated because it results in a decrease in the CPP of the brain and can also cause local compression of brain tissue against the tentorium, falx, and foramen magnum and ultimately herniation.3
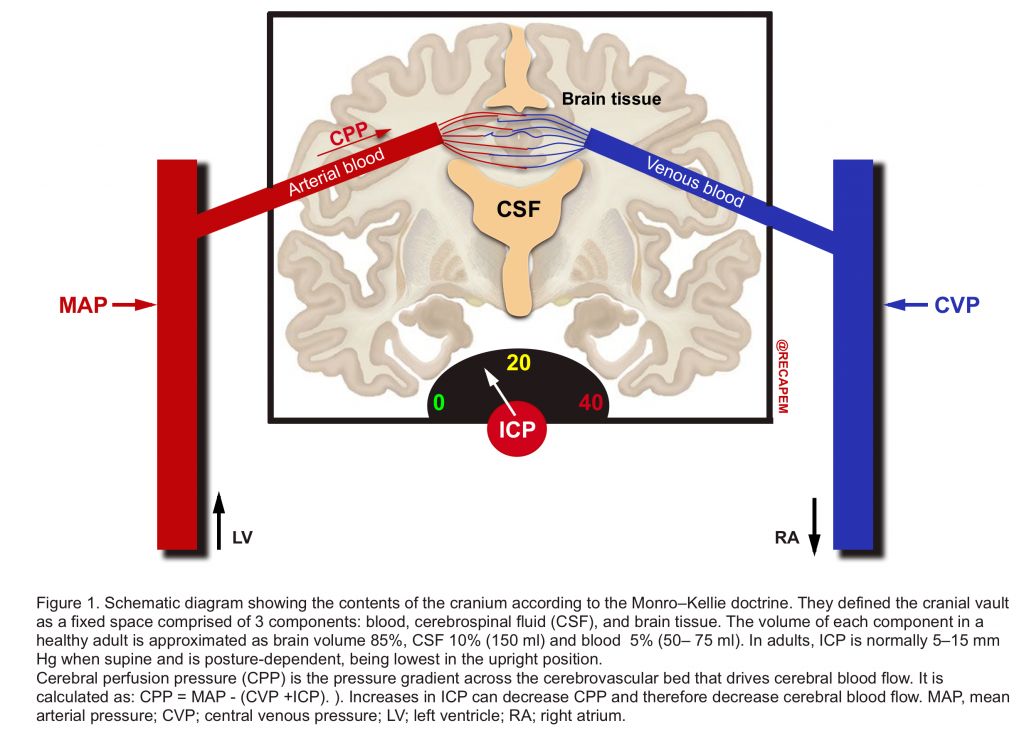
- Although the brain constitutes only 2% of body mass (1400 g), it receives a large proportion (12–15%) of the resting cardiac output in the adult. CBF has a direct relationship with cerebral perfusion pressure and has an inverse relationship with cerebral vascular resistance (CVR).
| CBF = CPP➗ CVR Whereas CPP = MAP – (CVP +ICP) CPP, cerebral perfusion pressure; CBF, cerebral blood flow; CVP, central venous pressure; CVR; cerebral vascular resistance; ICP, intracranial pressure; MAP, mean arterial pressure. |
- Thus, CBF will augment if the CPP increases and the cerebral vasculature is vasodilated (↓CVR). 2 4
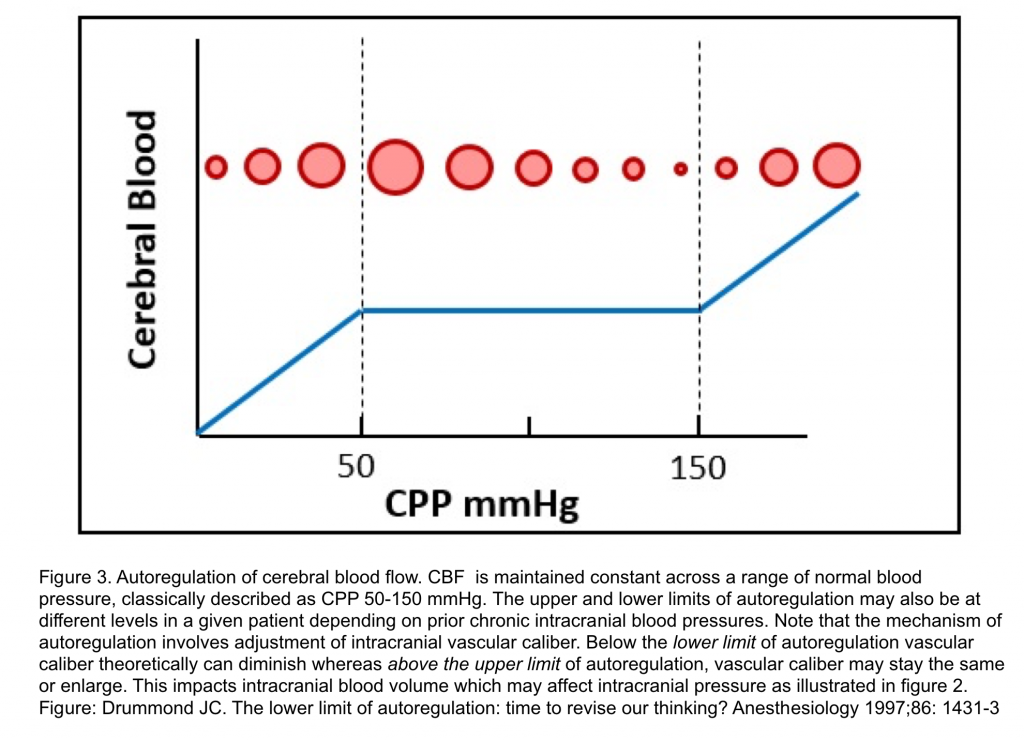
- Autoregulation of CBF
- Refers to the property of the brain to maintain relatively constant blood flow and oxygen supply in the face of changes in CPP.
- In normotensive adults with intact autoregulation, CBF is maintained at a constant rate of about 50 mL/min/100 g when CPP varies within a classically described range of 50 to 150 mmHg 2.
- Mechanism of CBF autoregulation
- Myogenic autoregulation is change in cerebrovascular resistance by vasoconstriction and vasodilation in response to changes in CPP.
- With intact autoregulation, when a decrement in CPP approaches the lower limit of autoregulation, vasodilation occurs to maintain CBF but, as CPP decreases below the lower limit of autoregulation, maximum cerebral vasodilation arises.
- This initially associates with a proportional CBF decrement as oxygen extraction increases to meet metabolic demand as a secondary compensation. However, further CBF decrease produces anaerobic cerebral ischemia and, ultimately, infarction.
- Conversely, ↑CPP within the autoregulatory range induces cerebral vasoconstriction. When autoregulation is intact, an increase in CPP above its upper limit produces dysregulated elevated CBF 5.
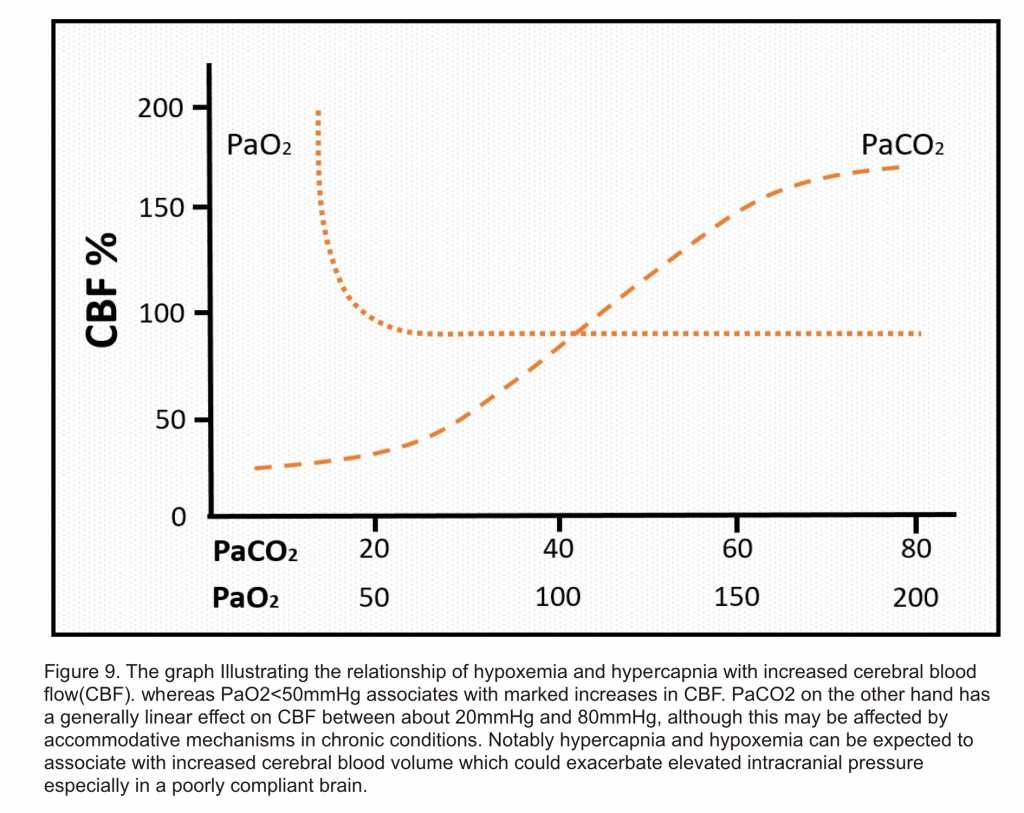
- Other parameters which can affect vascular tone and therefore contribute to vasodilation and hyperemia include metabolic factors (e.g. hypercapnia, hypoxemia, anemia, hypoglycemia) and neuronal activation (e.g. seizure, fever, noxious stimuli(e.g. Suctioning, endotracheal intubation, paroxysmal sympathetic hyperactivity)5.
Cerebral perfusion pressure(CPP)
- Perfusion pressure is the difference in the pressures between the arterial and venous circulation which dictates the blood flow to the organ. In the brain, the perfusion pressure or the CPP is affected by another pressure within the skull i.e. intracranial pressure.
- CPP= MAP – (CVP+ICP)
- In normal adults, the CPP is variable, usually ranging between 70 and 90 mm Hg and the CBF is constant.
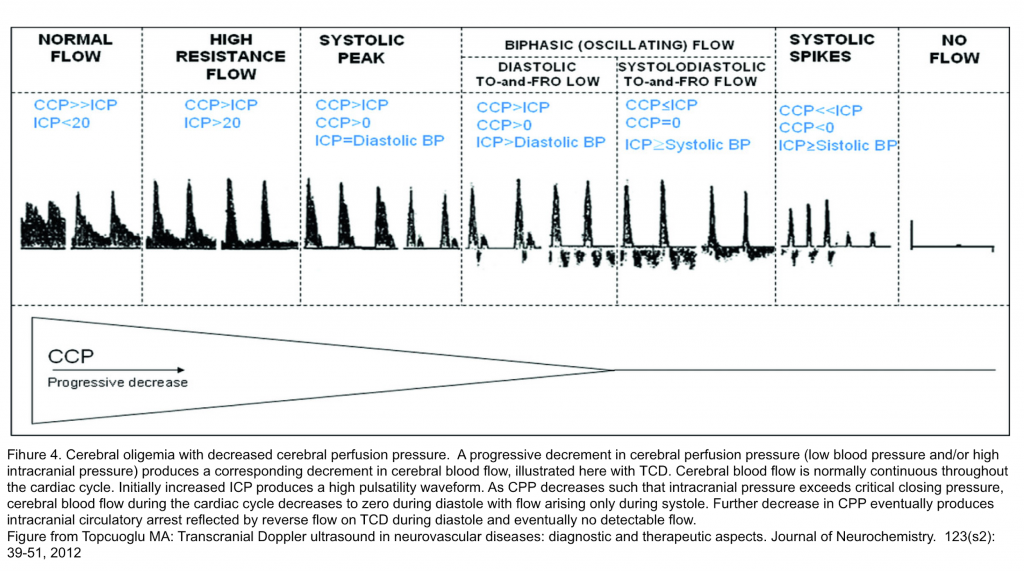
- When the CPP decreases below 50 mm Hg, there is an increased risk of brain ischaemia.
- In pathological conditions, if the ICP is increased, the flow through the cerebral blood vessels can be restricted. If ICP increases to the level of critical closing pressure, CBF, ordinarily continuous throughout the cardiac cycle, becomes discontinuous and there is no flow during diastole. If ICP exceeds systolic blood pressure, intracranial circulatory arrest may occur (Figure 4).5
Primary and secondary brain injury
- Primary brain injury is the direct result of a disease or an injury (neurotrauma) to the brain tissue.
- Secondary brain damage is not a simple condition but is a general term for secondary brain injury that occurs as a result of primary brain injury.1
- The main pathologies of secondary brain injury include elevated ICP, brain hypermetabolism, neuroinflammation, and secondary damage to areas surrounding brain damage (figure 5).
- Depending on the severity, and type of the primary insult, the brain tissue is vulnerable and is exposed to variable degree and extent of a secondary hit prompted by a series of molecular cascade events.
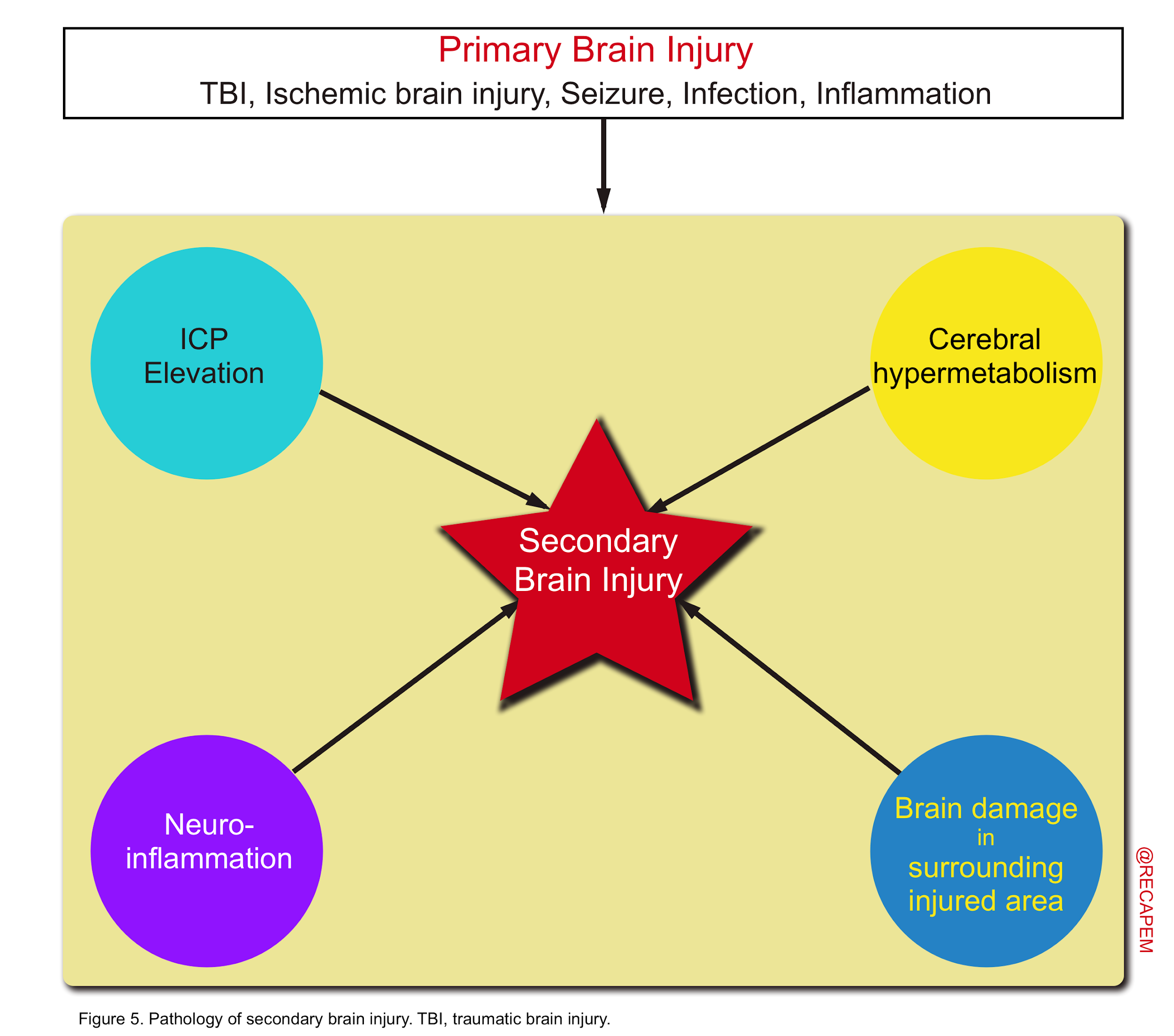
Pathophysiology of secondary brain injury
ICP elevation
Increased intracranial pressure can be a surrogate of secondary brain injury, as high ICP has been correlated with high mortality and poor neurological outcome 6. ICP monitoring and control have thus been the cornerstones of the neurocritical care management of brain-injured patients.
ICP monitoring: Methods for ICP monitoring can be divided into invasive and non-invasive approaches.
- Invasive methods include fluid-based systems and implantable micro-transducers. Of the invasive methods, ICP monitoring using an EVD is considered as the gold standard (figure 6), not only for its accuracy but also because it additionally serves a therapeutic purpose by allowing CSF drainage. ICP waveform is shown below (figure7).3
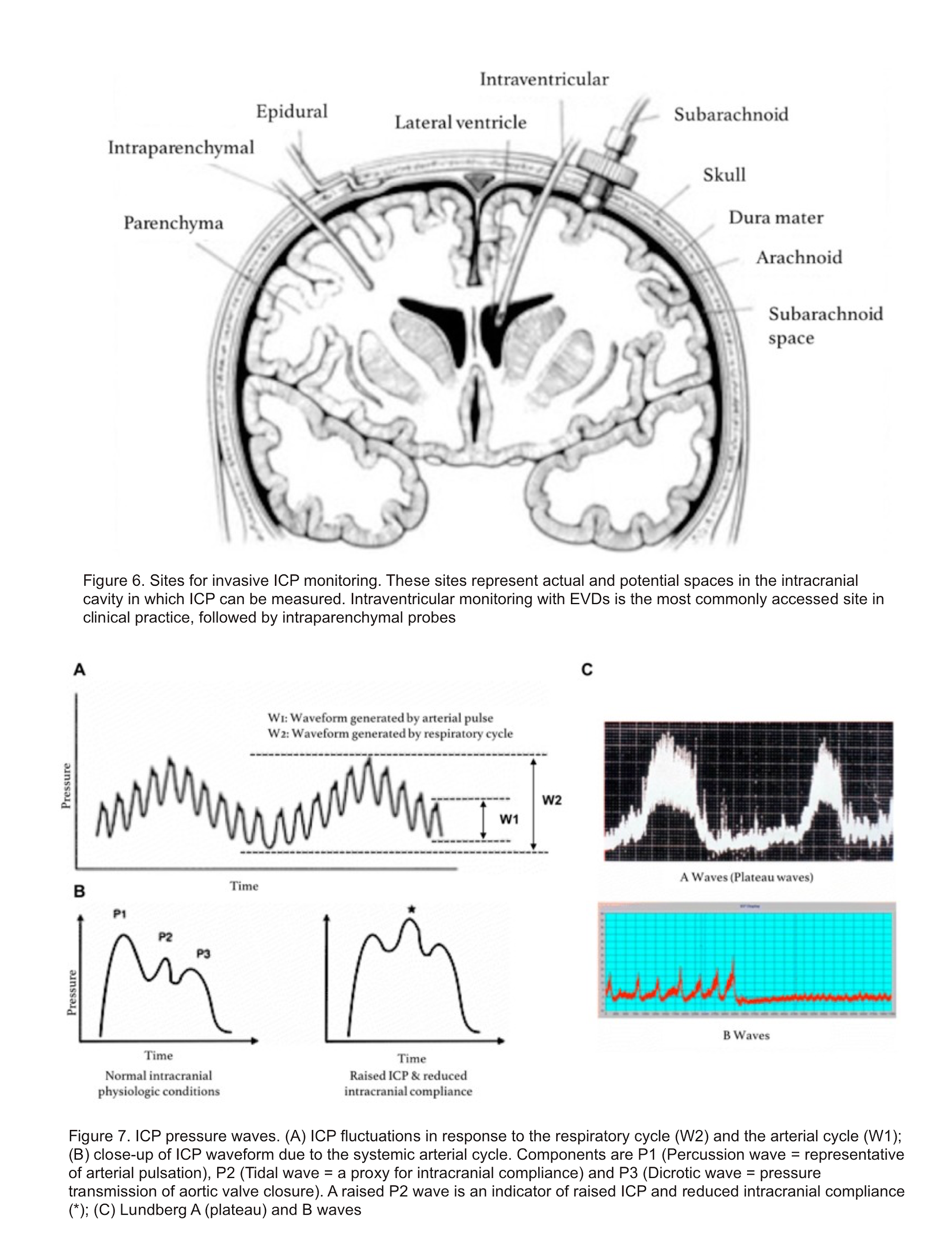
- Non-invasive method: Invasive method is the gold standard for assessing ICP. However it is not always feasible. Non-invasive methods can be used to screen patients for elevated ICP in situations where invasive interventions cannot be promptly accessed, such as in the field or where there are no neurosurgeons.Additionally, non-invasive screening can be done in patients in whom there is relatively low suspicion of elevated ICP, but the possibility needs to be ruled out. This may decrease the placement of invasive monitors in patients who, in retrospect, did not need them.3
- Transcranial Doppler (TCD)
- In the neuro-critical setting, TCD is most commonly used as a tool to monitor changes in cerebral blood flow (CBF) in the setting of subarachnoid hemorrhage-associated vasospasm. A number of models using TCD-derived data have shown correlation with invasively-measured ICP; these models have used measurements of flow velocity (FV) in the middle cerebral artery, arterial blood pressure and pulsatility index (PI) 7. This technology may be incorporated into clinical practice sooner as a neuro-monitoring adjunct rather than an ICP sensor 8.
- Optic Nerve Sheath Diameter (ONSD)
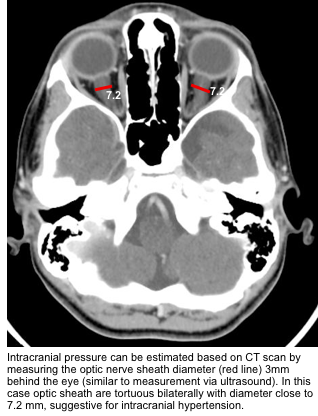
- Several studies have demonstrated a correlation between invasively measured ICP and ultrasonographic ONSD measurements, with overall sensitivity and specificity of 0.90 and 0.85, respectively. (watch media here).
- When measured 3 mm behind the eye:
- <5 mm is normal.
- 5-6 mm is a grey zone.
- >6 mm suggests abnormal ICP.
- Transcranial Doppler (TCD)
- Imaging-Based Methods including CT, MRI
- There are a variety of gross anatomic changes associated with elevated ICP that can be detected using CT and MRI.
- Optic nerve sheath diameter can be measure 3mm behind the globe (similar to measurement by ultrasound).
- For instance, the presence of a mass occupying lesion can cause compression of the ventricles and midline shift.
- Similarly, enlarged ventricles can be indicative of hydrocephalus, and cerebral edema can cause a loss of differentiation of grey and white matter junctions 10. While imaging continues to be used qualitatively, at present, these methods are not independently reliable enough as screening tools for elevated ICP 11.
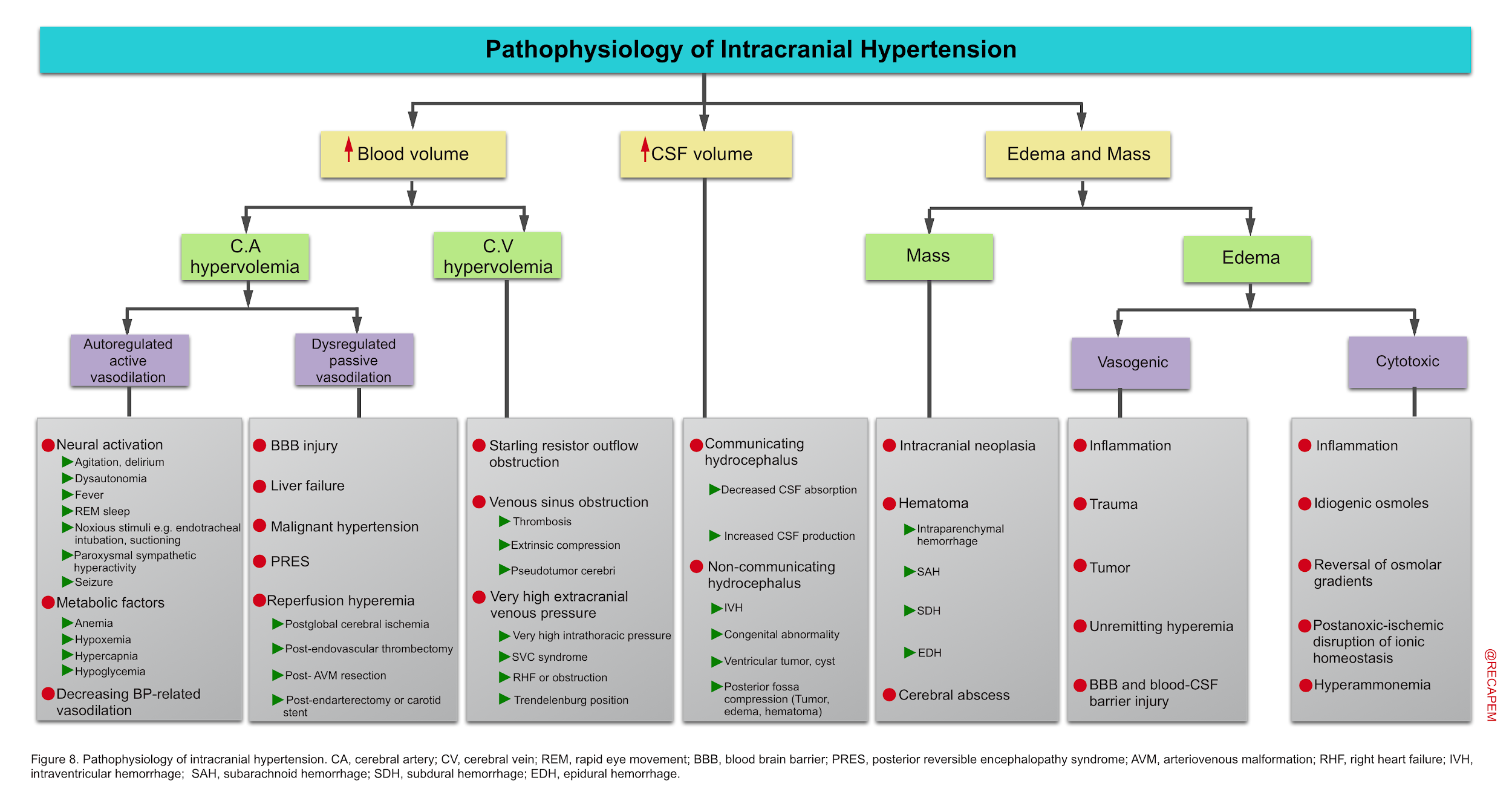
- Interpretation of elevated ICP is not straightforward as multiple dissimilar pathophysiological processes may concurrently contribute to the observed numerical ICP value.
- Arguably, etiologic contributors to elevated ICP and its physiological consequences should be major factors in developing rational therapeutic strategies.
- Treatment strategies used in one cause of ICP elevation, for example hyperemia, may be ineffective or even harmful if applied in others, for example edema producing ischemia, or hydrocephalus.
- Therefore a more individualized approach to intracranial hypertension based on pathophysiologic processes is deserved 5.
- Intracranial hypertension can be caused by ↑blood volume, ↑brain tissue volume (masses, and edema), and ↑cerebrospinal fluid (figure below).
- While these pathophysiological subsets provide a conceptual framework for approaching intracranial hypertension, it is important to be cognizant these processes seldom arise in isolation, but often in combination.
- For example, a hyperemic process may initially increase ICP but then contribute to subsequent edema or hemorrhage with further increases in ICP, but now with oligemia.
1.Increased blood volume
As explained above, in the normal state, physiological increases in cerebral blood volume do not increase ICP unless the compensatory capacitive mechanisms were exhausted. Increased intravascular blood volume can be related to cerebral arterial and cerebral venous hypervolemia.
- Cerebral arterial hypervolemia: Arterial vasodilation can happen in the face of preserved cerebral autoregulation wherein metabolic and neurogenic factors can cause active vasodilation; or when autoregulatory mechanism is disrupted such as in malignant hypertension. In the latter situation, passive arterial vasodilation can contribute to cerebral arterial hypervolemia.
-
- Active vasodilation in the presence of intact autoregulatory mechanism
- Neuronal activation: Conditions of abnormally increased metabolic demand can increase CBF, with an ICP elevation if compliance is abnormal. These include increased:
- Metabolic mediators: Hypercapnia 17, hypoxemia 18,hypoglycemia 19, and anemia 20, are all normally associated with hyperemic responses linked to maintenance of nutrient supply and removal of waste products from the brain (figure 9). These physiological responses to metabolic mediators associated with hyperemia are normally well tolerated, but can increase ICP in certain situations such as a poorly compliant brain in the case of neurotrauma.
- Active vasodilation in the presence of intact autoregulatory mechanism
-
-
- Decreasing Blood Pressure-related Vasodilation
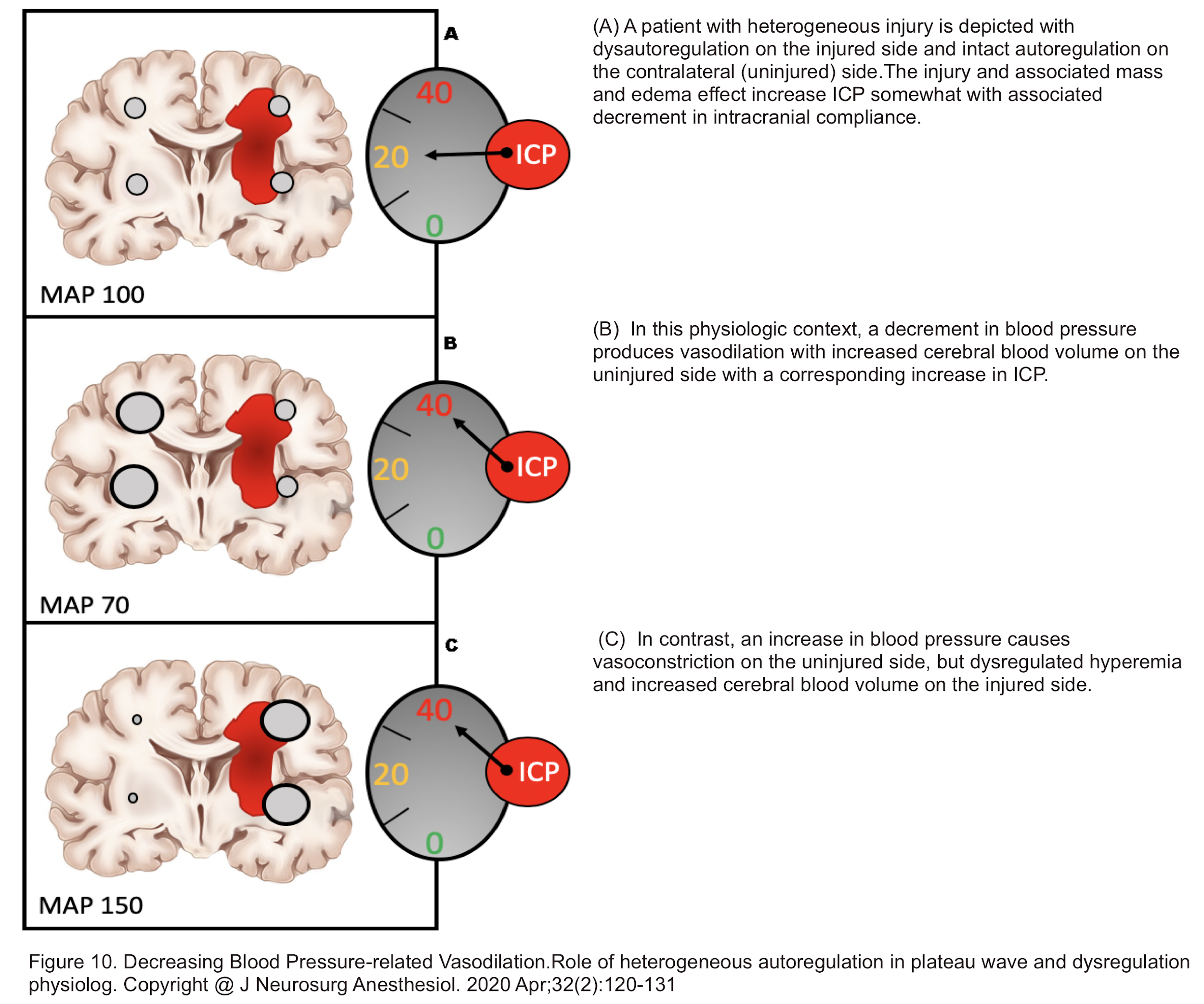
- In the presence of heterogeneous autoregulation wherein a traumatic brain injury has caused dysautoregulation in the corresponding side, whereas the intact part of the brain is functioning normally (intact autoregulation); a drop in blood pressure can cause ICP elevation (figure 10) 21.
- Decreasing Blood Pressure-related Vasodilation
-
-
- Dysregulated Passive Arterial Vasodilation
- Encompasses conditions wherein primarily the cerebral autoregulation of blood flow is disrupted resulting in dilated poorly reactive vascular beds and capillary leakage. These conditions are associated with endothelial and BBB injury and can be caused by:
- Brain injury (ischemic or traumatic) 22
- Acute liver failure 23
- Hyperperfusion states such as malignant hypertension, PRES 24 25
- Arteriovenous malformation (AVM) resection with subsequent normal perfusion pressure breakthrough 26
- Post carotid endarterectomy 27 or endovascular thrombectomy with successful restoration of CBF 28.
- In addition, secondary disruption of autoregulation can arise in extremes of hypercapnia, hypoxemia, anemia, hypotension, and hypoglycemia, by which brain edema may be exacerbated.
- In all of these cases of extreme vasodilation, CBF is expected to vary directly with systemic blood pressure 5.
- Encompasses conditions wherein primarily the cerebral autoregulation of blood flow is disrupted resulting in dilated poorly reactive vascular beds and capillary leakage. These conditions are associated with endothelial and BBB injury and can be caused by:
- Dysregulated Passive Arterial Vasodilation
- Cerebral venous hyperemia
- Increased cerebral venous or dural venous sinus pressures may increase brain tissue hydrostatic pressure and thereby contribute to vasogenic edema 5. Primary factors contributing to ↑cerebral venous volume include:
- Functional starling resistor type venous outflow obstruction
- Increased ICP, from whatever etiology, can also produce a direct constriction of venous outflow at the level of thin-walled cerebral veins. This venous obstruction produces an increase in internal venous pressure, which maintains vein patency such that flow continues. However, it also increases tissue hydrostatic pressure, which then presents the potential to produce or exacerbate cerebral edema which further increases ICP with a positive feedback cycle thus produced 29 30.
- The Starling resistor concept, sometimes referred to as the vascular waterfall phenomenon, describes the situation where external tissue pressure is greater than internal vascular pressure. In such a situation hollow tubes such as blood vessels collapse and flow then become dependent on the difference between proximal upstream pressure and distal external extravascular pressure at the point of constriction.
- Increased ICP, from whatever etiology, can also produce a direct constriction of venous outflow at the level of thin-walled cerebral veins. This venous obstruction produces an increase in internal venous pressure, which maintains vein patency such that flow continues. However, it also increases tissue hydrostatic pressure, which then presents the potential to produce or exacerbate cerebral edema which further increases ICP with a positive feedback cycle thus produced 29 30.
- Mechanical outflow obstruction, such as with venous thrombosis 31 32
- Given that CSF reabsorption occurs at arachnoid granulations in the cerebral venous sinuses, thrombosis of dural sinuses can impair CSF reabsorption resulting in increased CSF volume. This can cause communicating hydrocephalus and increased ICP.
- Cerebral venous sinus thrombosis is believed to produce an increase in local venular and capillary pressures from venous outflow obstruction. This produces or exacerbates BBB disruption with consequent vasogenic edema and oligemia/ischemia.
- Venular and capillary disruption can also result in intraparenchymal hemorrhage to increase ICP. If the hemorrhage includes the ventricular system, the process may be further complicated by noncommunicating hydrocephalus.
- Vasogenic edema with worsened intracranial hypertension can result in further venous outflow obstruction by a Starling resistor effect, further exacerbating the pathophysiological process.
- Very high extracranial venous press
- Anything that increases venous pressure sufficient to fall within the range of ICP at the level of the cranium carries a theoretical risk of increased intracranial venous volume. Such situations may include
- High airway pressure 33
- Acute superior vena cava syndrome
- Right heart failure
- Trendelenburg head-down position
- Anything that increases venous pressure sufficient to fall within the range of ICP at the level of the cranium carries a theoretical risk of increased intracranial venous volume. Such situations may include
- Functional starling resistor type venous outflow obstruction
- Increased cerebral venous or dural venous sinus pressures may increase brain tissue hydrostatic pressure and thereby contribute to vasogenic edema 5. Primary factors contributing to ↑cerebral venous volume include:
2.Mass and edema
- In contrast to increased intravascular blood volume subsets of elevated ICP, masses and edema can directly produce oligemic intracranial hypertension and have immediate deleterious effects.
- Brain tissue edema: Diffuse brain tissue edema is an important cause of intracranial hypertension and is commonly entwined with other causes of increased ICP. Brain edema is an abnormal accumulation of fluid within the brain parenchyma and is subdivided into two major categories; vasogenic and cytotoxic edema.34 35
- Vasogenic edema: It encompasses conditions associated with BBB breakdown, allowing movement of intravascular proteins, solutes, and water through the damaged microvascular endothelial cells into the extracellular space.
- Etiologies of vasogenic edema are numerous and include
- Traumatic brain injury
- Brain tumor
- Radiation necrosis
- Acute demyelination
- Dysregulated hyperemia
- Venous obstruction
- Inflammatory/infectious process.
- When brain edema overcomes the compensatory mechanisms, ICP, following an exponential relationship with volume of an intracranial compartment, can increase dramatically resulting in compromise of CBF with widespread ischemia or in herniation (figure 2 above) 36.
- Etiologies of vasogenic edema are numerous and include
- Cytotoxic edema: It develops following processes that arise and cause intracellular fluid translocation. In the case of hypoxia-ischemia, a primary insult leads to failure of ATP-dependent sodium-potassium and calcium pumps resulting in intracellular sodium (or other metabolite) accumulation, an osmotic gradient, fluid shift from extracellular to intracellular compartments, and eventual cellular swelling. Other syndromes may increase different intracellular osmoles to produce the same effect. Calcium can also accumulate within the cell, triggering an inflammatory cascade that recruits microglia and leads to free radical formation that ultimately results in the destruction of the BBB and concurrent vasogenic edema.37
- Causes of cytotoxic edema include:
- Vasogenic edema: It encompasses conditions associated with BBB breakdown, allowing movement of intravascular proteins, solutes, and water through the damaged microvascular endothelial cells into the extracellular space.
- Masses: Pathologic intracranial masses can increase ICP, eventually culminating in oligemia.
- Masses can be in the form of hematoma or neoplasm, abscess.
- The more rapid the growth of said mass, the more acute and profound the rise in ICP.
- Brain tissue edema: Diffuse brain tissue edema is an important cause of intracranial hypertension and is commonly entwined with other causes of increased ICP. Brain edema is an abnormal accumulation of fluid within the brain parenchyma and is subdivided into two major categories; vasogenic and cytotoxic edema.34 35
3.Increased CSF volume
- CSF Dynamic
- CSF is produced mainly by epithelial cells of the choroid plexus lining the cerebral ventricles using active transport, largely dependent on the enzyme carbonic anhydrase.
- CSF then circulates uni-directionally through the ventricular system, from the lateral ventricles to the foramen of Monro, into the third ventricle, aqueduct of Sylvius, the fourth ventricle, and finally exiting into the spinal subarachnoid space through the foramina of Luschka and Magendie.
- The spinal subarachnoid space is in direct communication with the intracranial subarachnoid space from where CSF is passively absorbed down a pressure gradient through the arachnoid villi of dural venous sinuses into venous sinuses 40.
- In order for this process to occur, CSF pressure must exceed sagittal sinus venous pressure.
- From the venous sinuses, CSF eventually enters into the systemic venous circulation.
- This classic description of CSF dynamics has recently been enhanced to incorporate the glymphatic system, a perivascular intraparenchymal pathway for CSF egress from the brain 41.
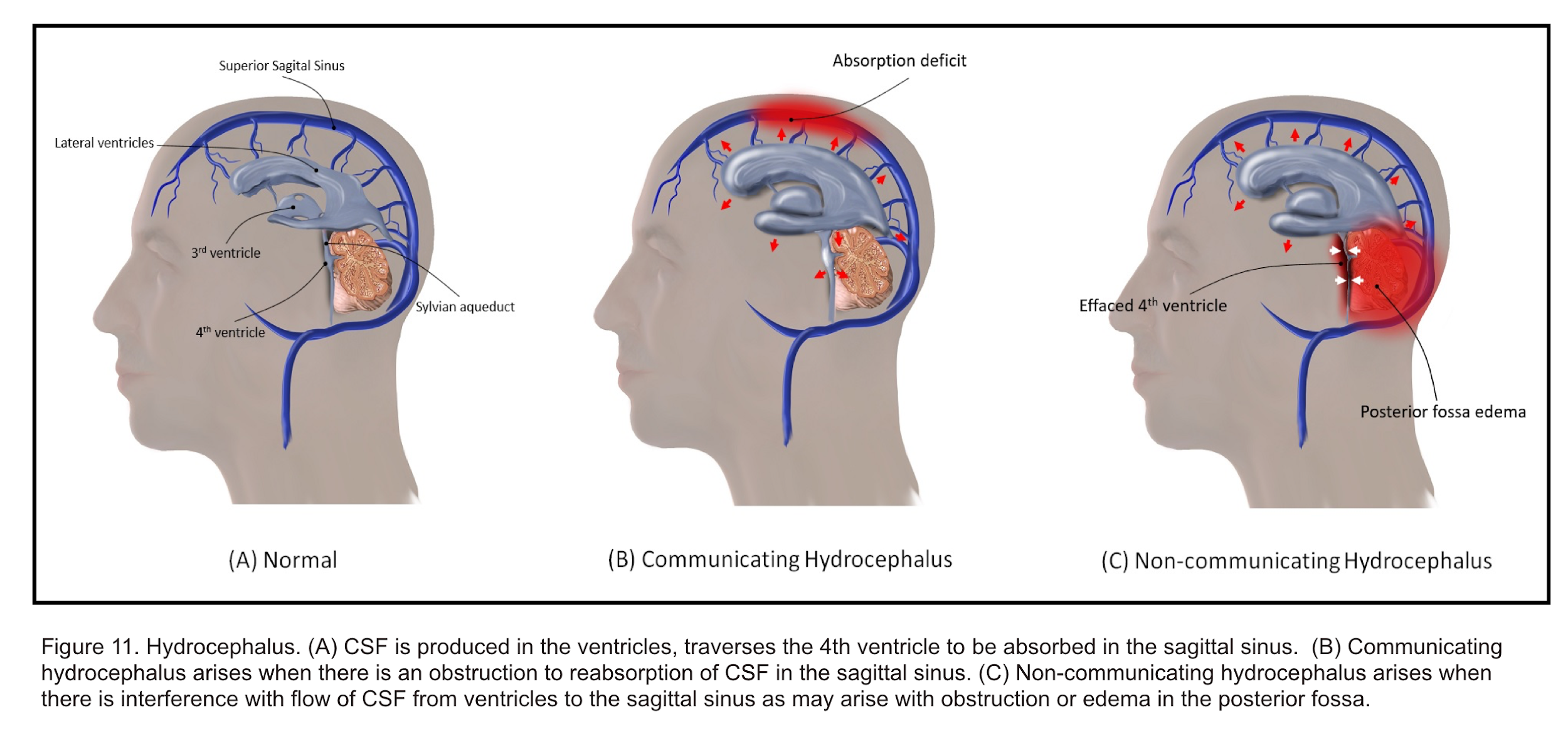
- Hydrocephalus
- It is a disruption of CSF formation, flow, or absorption characterized by excessive accumulation of CSF within the cerebral ventricular system and/or subarachnoid space. Regardless of cause, this can result in ventriculomegaly and intracranial hypertension. Hydrocephalus was historically classified by Dandy into 2 types; communicating and non-communicating.42
- Communicating hydrocephalus
- Occurs due to excess CSF production or, more commonly, ineffective CSF absorption. For example, it can be a result of scarring or fibrosis in the subarachnoid space after infection, inflammation, or hemorrhage.
- Non-communicating hydrocephalus
- Occurs from obstruction of CSF flow within the ventricular system or its outlets, resulting in dilation of the ventricular system proximal to the point of obstruction.
Cerebral hypermetabolism
- The brain consumes 20% of the body’s total oxygen supply. Brain metabolism is dependent on glucose and oxygen transported by the blood.
- In neurological emergencies (e.g. head trauma and stroke, etc) the blood supply does not fit to the demand and this will cause relative ischemia and anaerobic metabolism in which lactic acid is produced , and secondary brain damage is promoted.
- This condition leads to further worsening of brain damage when accompanied by convulsive seizures, systemic hypoxia, decreased CPP due to reduced blood pressure, reduced oxygen-carrying capacity due to decreased hemoglobin, and the onset of other factors.
- Hyperglycemia is also known to promote secondary brain damage.
- It promotes anaerobic glycolysis that causes lactic acid accumulation, resulting in lactic acidosis, which is considered a cause of aggravated secondary brain damage.
Neuroinflammation
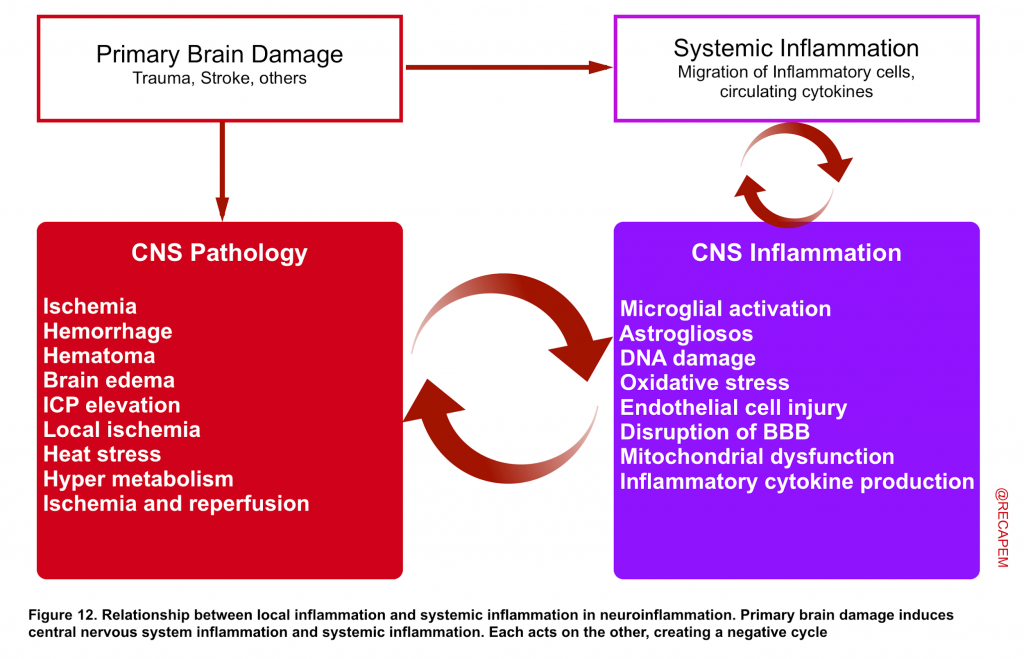
- Blood cells do not naturally reside within the brain tissue. Cerebral blood vessels are separated from the brain by the blood–brain barrier (BBB) and are normally isolated from the systemic immune system 43 44.
- Disruption of the BBB has been discovered to be involved in different primary brain injuries such as neurotrauma, stroke, and Alzheimer’s disease 45. The vascular endothelial dysfunction and dysfunction of nearby astrocytes in the cerebral blood vessels surrounding the site of injury cause the tight junctions of the BBB to break down, resulting in disruption of the BBB. This results in the so-called neuroinflammation, in which negative cross talk occurs between local inflammation in the brain and systemic inflammation 46.
- Almost immediately after the occurrence of a brain injury, microglia are activated and produce active oxygen and inflammatory cytokines, which engulf nerve cells and other cells that can cause cell death 47.
- In addition to macrophages migrating to the brain and acting as inflammatory cells due to disruption of the BBB, circulating inflammatory cytokines in the body penetrating the BBB to act on the brain are one of the many important actions resulting in the spread of systemic inflammation to the brain 48. This promotes an inflammatory response in the damaged part of the brain and exacerbates secondary brain injury 49.
- This inflammatory response is mediated by multiple different molecular cascade events, which is beyond the scope of this discussion. Of note, hyperglycemia caused by disease-related stress can also exacerbate neuro-inflammation 50 51.
Secondary brain damage in the surrounding injured area
Many of the aforementioned secondary brain damage pathologies will expand to involve adjacent areas of the primary brain damage which if not prevented and suppressed leads to higher brain dysfunction e.g. paralysis.
- In particular, the penumbra area in ischemic stroke, an area of decreased blood flow, exists from the start surrounding the area of brain injury, which causes irreversible dysfunction when secondary brain damage develops in this area.
- Thus, acute treatment through neurocritical care is vital regardless of the severity of injury 52.
Treatment of secondary brain injury
- As mentioned earlier, different types of pathologies (figure 5) that contribute to secondary brain injury warrants different treatment. The various pathways of cellular injury have been the focus of extensive preclinical work into the development of neuroprotective treatments to prevent secondary brain injury. At this time; no clinical trials of these strategies have demonstrated clear benefit in patients.
- However, a critical aspect of ameliorating secondary brain injury is the avoidance of secondary brain insults (e.g. optimizing CPP, avoiding hypotension, hypoxia, hyperglycemia, etc) and resolution of the primary brain injury (e.g. evacuation of a blood clot, resection of a tumor, CSF diversion in the setting of hydrocephalus, or treatment of an underlying metabolic disorder).
- The emergent assessment and support of the airway, oxygenation, organ perfusion are applicable to all patients.
- If intubation is indicated, resuscitate before intubate.
- Optimize BP, arterial oxygenation before proceeding to intubation as hypotension, especially in conjunction with hypoxemia, can induce reactive vasodilation and elevations in ICP.
- There’s no good clinical evidence supporting the use of systemic lidocaine to prevent ICP rise before intubation. Topical lidocaine is more effective than intravenous routes.
- For full lecture on this visit: The Neurocritical Care Intubation(EMCrit RACC)
Neuroresuscitation
- Elevated ICP is not a homogenous entity and each different pathophysiologic mechanisms responsible for ICP elevation may require a unique treatment strategy.
- Keep in mind that the treatment goal is to protect cerebral perfusion pressure (figure1) and the only way to reliably determine CPP is to continuously monitor both ICP and blood pressure.
- Therefore, while initial empiric therapy to control presumed elevated ICP may be performed without the benefit of ICP monitoring, an important early goal in management of the patient with presumed elevated ICP is close monitoring of both ICP (via placement of an ICP monitoring device) and BP (via arterial line).
- Elevated ICP is a medical emergency and treatment should be undertaken promptly. In addition to definitive treatment, there are measures that can be employed to reduce ICP acutely (though their therapeutic effect might be transient).
- Brain edema
- Head-up position
- Elevation of the head to at least 30° is advised in patients with raised ICP, with one study confirming that this degree of elevation produces consistent reduction in ICP 53 (moderate elevation is safe as long as CPP is continuously maintained at > 60 mm Hg).
- Osmotic therapy and diuresis
- May include hypertonic saline, mannitol and furosemide.
- Further trials are required to show superiority of hypertonic saline over mannitol.
- Head-up position
- Control of cerebral blood volume
- Cerebral blood flow control
- Hyperventilation: Use of mechanical ventilation to lower PaCO2 to 26 to 30 mmHg has been shown to rapidly reduce ICP through vasoconstriction and a decrease in the volume of intracranial blood.
- It can be an effective urgent intervention when elevated ICP complicates cerebral edema, intracranial hemorrhage, and tumor.
- It should Not be used in the setting of TBI, or acute stroke, wherein vasoconstriction may cause a critical decrease in local cerebral perfusion and worsen neurologic injury, particularly in the first 24 to 48 hours 54.
- It should be emphasized that regardless of the cause of elevated ICP, hyperventilation has a short-lived effect (1 to 24h), and Following therapeutic hyperventilation, the patient’s respiratory rate should be tapered back to normal over several hours to avoid a rebound effect 55.
- Blood pressure control: Both extreme hypotension and hypertension can impair cerebral perfusion pressure especially in neurologically vulnerable patients.
- By enlarge, BP should be controlled sufficient to keep 60 < CPP > 120 mm Hg.
- Hyperventilation: Use of mechanical ventilation to lower PaCO2 to 26 to 30 mmHg has been shown to rapidly reduce ICP through vasoconstriction and a decrease in the volume of intracranial blood.
- Prevention of venous congestion
- Head-up position
- Control of intrathoracic pressure
- Avoidance of compression of the jugular vein
- Cerebral blood flow control
- Control of CSF
- CSF drainage: When hydrocephalus is identified, a ventriculostomy should be inserted.
- When indicated, CSF should be “slowly” removed at a rate of approximately 1 to 2 mL/minute, for 2-3 minutes at a time, with intervals of 2-3 minutes in between until a satisfactory ICP has been achieved (ICP <20 mmHg) or until CSF is no longer easily obtained.
- CSF drainage: When hydrocephalus is identified, a ventriculostomy should be inserted.
- Control of mass effect
- Control of brain edema
- Coagulopathy: in the presence of life threatening hemorrhage, coagulopathy should be corrected.
- Operation (internal decompression, external decompression).
- Brain edema
B.Management of cerebral hypermetabolism
- Fever
- Fever increases brain metabolism. Elevated metabolic demand in the brain results in increased cerebral blood flow and can elevate ICP by increasing the volume of blood in the cranial vault.
- Acetaminophen and cooling blankets are the first line of therapy and should be instituted when temperature is sustained > 38.3°C 59.
- Seizure
- Seizures can both complicate and contribute to elevated ICP.
- Anticonvulsant therapy should be instituted if seizures are suspected; prophylactic treatment may be warranted in some cases.
- There are no clear guidelines for prophylactic use, but examples include high-risk mass lesions, such as
- Supratentorial cortical mass locations
- Lesions adjacent to the cortex, such as subdural hematomas or subarachnoid hemorrhage.
- Agitation
- Agitation must be avoided because it can aggravate ICP elevation through straining (increasing thoracic, jugular venous, and systemic blood pressure) and increased brain metabolic rate.
- For this reason, adequate sedation must be the first pharmacologic intervention in managing an ICP crisis.
- Preferred agents are short acting opioids such as fentanyl (1-3 μg/kg/h) to provide analgesia and propofol (0.3-3 mg/kg/h).
- Compared with an opioid-based sedation regimen, in one trial propofol was associated with lower ICP and fewer ICP interventions in patients with severe TBI 60.
C.Prevention of neuro-inflammation
- Prevention of hyperglycemia
- Control of secondary systemic inflammatory response (infection, others)
- Other theoretical potential targets for therapy in future may include:
- Hypothermia, measures for prevention of BBB disruption, and endothelial cell injury, suppression of glial activation, suppression of inflammatory agents production (free radicals, HMGB1, inflammatory cytokines) and antioxidant therapy.
D.Prevention of secondary brain damage in surrounding injured area
- The basic views on secondary brain injury localized to the area of brain injury correspond to the aforementioned above points
Media
Going further
- EM:RAP HD Video: Cranial Burr Hole
- Elevated intracranial pressure (IBCC)
- Analgesia for the critically ill patient (IBCC)
- Traumatic Brain Injury (IBCC)
- Post-craniotomy management and complications (IBCC)
- NeuroCritical Care with Neha Dangayach (EMCrit)
References
1. Kaur P, Sharma S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr Neuropharmacol. 2018;16(8):1224-1238. doi: 10.2174/1570159X15666170613083606. PMID: 28606040
2. Alifia Tameem, MBBS MD FRCA, Hari Krovvidi, MD FRCA, Cerebral physiology, Continuing Education in Anaesthesia Critical Care & Pain, Volume 13, Issue 4, August 2013, Pages 113–118, https://doi.org/10.1093/bjaceaccp/mkt001
3. Harary M, Dolmans RGF, Gormley WB. Intracranial Pressure Monitoring-Review and Avenues for Development. Sensors (Basel). 2018;18(2):465. Published 2018 Feb 5. doi:10.3390/s18020465
4. Donnelly J, Budohoski KP, Smielewski P, Czosnyka M. Regulation of the cerebral circulation: bedside assessment and clinical implications. Crit Care. 2016;20(1):129. Published 2016 May 5. doi:10.1186/s13054-016-1293-6
5. Kofke WA, Rajagopalan S, Ayubcha D, Balu R, Cruz-Navarro J, Manatpon P, Mahanna-Gabrielli E. Defining a Taxonomy of Intracranial Hypertension: Is ICP More Than Just a Number? J Neurosurg Anesthesiol. 2020 Apr;32(2):120-131. doi: 10.1097/ANA.0000000000000609. PMID: 31135572; PMCID: PMC6872925
6. Miller JD, Becker DP, Ward JD, Sullivan HG, Adams WE, Rosner MJ. Significance of intracranial hypertension in severe head injury. J Neurosurg. 1977 Oct;47(4):503-16. doi: 10.3171/jns.1977.47.4.0503. PMID: 903804
7. Rasulo FA, Bertuetti R, Robba C, Lusenti F, Cantoni A, Bernini M, Girardini A, Calza S, Piva S, Fagoni N, Latronico N. The accuracy of transcranial Doppler in excluding intracranial hypertension following acute brain injury: a multicenter prospective pilot study. Crit Care. 2017 Feb 27;21(1):44. doi: 10.1186/s13054-017-1632-2. PMID: 28241847; PMCID: PMC5329967
8. Willie CK, Colino FL, Bailey DM, Tzeng YC, Binsted G, Jones LW, Haykowsky MJ, Bellapart J, Ogoh S, Smith KJ, Smirl JD, Day TA, Lucas SJ, Eller LK, Ainslie PN. Utility of transcranial Doppler ultrasound for the integrative assessment of cerebrovascular function. J Neurosci Methods. 2011 Mar 30;196(2):221-37. doi: 10.1016/j.jneumeth.2011.01.011. Epub 2011 Jan 27. PMID: 21276818
9. Jeon JP, Lee SU, Kim SE, Kang SH, Yang JS, Choi HJ, Cho YJ, Ban SP, Byoun HS, Kim YS. Correlation of optic nerve sheath diameter with directly measured intracranial pressure in Korean adults using bedside ultrasonography. PLoS One. 2017 Sep 13;12(9):e0183170. doi: 10.1371/journal.pone.0183170. PMID: 28902893; PMCID: PMC5597178
10. Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002 May;33(5):1225-32. doi: 10.1161/01.str.0000015624.29071.1f. PMID: 11988595
11. Rebel PH, Bartek J Jr, Andresen M, Bellander BM, Romner B. Intracranial Pressure Monitoring: Invasive versus Non-Invasive Methods-A Review. Crit Care Res Pract. 2012;2012:950393. doi: 10.1155/2012/950393. Epub 2012 Jun 8. PMID: 22720148; PMCID: PMC3376474
12. Uchida D, Fujimoto A, Yamazoe T, Yamamoto T, Enoki H. Seizure frequency can be reduced by changing intracranial pressure: A case report in drug-resistant epilepsy. Epilepsy Behav Case Rep. 2018;10:14-17. Published 2018 Jan 10. doi:10.1016/j.ebcr.2017.12.005
13. Burney RG, Winn R. Increased cerebrospinal fluid pressure during laryngoscopy and intubation for induction of anesthesia. Anesth Analg. 1975 Sep-Oct;54(5):687-90. doi: 10.1213/00000539-197509000-00028. PMID: 1237257
14. Bein T, Kuhr LP, Bele S, Ploner F, Keyl C, Taeger K. Lung recruitment maneuver in patients with cerebral injury: effects on intracranial pressure and cerebral metabolism. Intensive Care Med. 2002 May;28(5):554-8. doi: 10.1007/s00134-002-1273-y. Epub 2002 Apr 12. PMID: 12029401
15. Nyholm L, Howells T, Lewén A, Hillered L, Enblad P. The influence of hyperthermia on intracranial pressure, cerebral oximetry and cerebral metabolism in traumatic brain injury. Ups J Med Sci. 2017;122(3):177-184. doi:10.1080/03009734.2017.1319440
16. Klug N, Hoffmann O, Zierski J, Buss K, Laun A, Agnoli AL. Decerebrate rigidity and vegetative signs in the acute midbrain syndrome with special regard to motor activity and intracranial pressure. Acta Neurochir (Wien). 1984;72(3-4):219-33. doi: 10.1007/BF01406872. PMID: 6475577
17. Sakabe T, Siesjö BK. The effect of indomethacin on the blood flow-metabolism couple in the brain under normal, hypercapnic and hypoxic conditions. Acta Physiol Scand. 1979 Nov;107(3):283-84. doi: 10.1111/j.1748-1716.1979.tb06476.x. PMID: 539458
18. Arbeláez AM, Su Y, Thomas JB, Hauch AC, Hershey T, Ances BM. Comparison of regional cerebral blood flow responses to hypoglycemia using pulsed arterial spin labeling and positron emission tomography. PLoS One. 2013;8(3):e60085. doi: 10.1371/journal.pone.0060085. Epub 2013 Mar 28. PMID: 23555895; PMCID: PMC3610825.
19. Floyd TF, McGarvey M, Ochroch EA, Cheung AT, Augoustides JA, Bavaria JE, Acker MA, Pochettino A, Detre JA. Perioperative changes in cerebral blood flow after cardiac surgery: influence of anemia and aging. Ann Thorac Surg. 2003 Dec;76(6):2037-42. doi: 10.1016/s0003-4975(03)01074-9. PMID: 14667637
20. LUNDBERG N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl. 1960;36(149):1-193. PMID: 13764297
21. Steiner LA, Czosnyka M, Piechnik SK, Smielewski P, Chatfield D, Menon DK, Pickard JD. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit Care Med. 2002 Apr;30(4):733-8. doi: 10.1097/00003246-200204000-00002. PMID: 11940737
22. Aggarwal S, Obrist W, Yonas H, Kramer D, Kang Y, Scott V, Planinsic R. Cerebral hemodynamic and metabolic profiles in fulminant hepatic failure: relationship to outcome. Liver Transpl. 2005 Nov;11(11):1353-60. doi: 10.1002/lt.20479. PMID: 16237715
23. Skinhoj E. On the pathogenesis of hypertensive encephalopathy as revealed by cerebral blood flow studies in man. Prog Brain Res. 1977;47:235-43. doi: 10.1016/S0079-6123(08)62728-4. PMID: 928747
24. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008 Jun;29(6):1043-9. doi: 10.3174/ajnr.A0929. Epub 2008 Apr 10. PMID: 18403560.
25. Alexander MD, Connolly ES, Meyers PM. Revisiting normal perfusion pressure breakthrough in light of hemorrhage-induced vasospasm. World J Radiol. 2010;2(6):230-232. doi:10.4329/wjr.v2.i6.230
26. Bouri S, Thapar A, Shalhoub J, Jayasooriya G, Fernando A, Franklin IJ, Davies AH. Hypertension and the post-carotid endarterectomy cerebral hyperperfusion syndrome. Eur J Vasc Endovasc Surg. 2011 Feb;41(2):229-37. doi: 10.1016/j.ejvs.2010.10.016. Epub 2010 Dec 4. PMID: 21131217
27. Goyal N, Tsivgoulis G, Pandhi A, Chang JJ, Dillard K, Ishfaq MF, Nearing K, Choudhri AF, Hoit D, Alexandrov AW, Arthur AS, Elijovich L, Alexandrov AV. Blood pressure levels post mechanical thrombectomy and outcomes in large vessel occlusion strokes. Neurology. 2017 Aug 8;89(6):540-547. doi: 10.1212/WNL.0000000000004184. Epub 2017 Jul 7. PMID: 28687721.
28. Grände PO, Asgeirsson B, Nordström CH. Volume-targeted therapy of increased intracranial pressure: the Lund concept unifies surgical and non-surgical treatments. Acta Anaesthesiol Scand. 2002 Sep;46(8):929-41. doi: 10.1034/j.1399-6576.2002.460802.x. PMID: 12190792.
29. Huseby JS, Luce JM, Cary JM, Pavlin EG, Butler J. Effects of positive end-expiratory pressure on intracranial pressure in dogs with intracranial hypertension. J Neurosurg. 1981 Nov;55(5):704-5. doi: 10.3171/jns.1981.55.5.0704. PMID: 7031199
30. Saposnik G, Barinagarrementeria F, Brown RD Jr, Bushnell CD, Cucchiara B, Cushman M, deVeber G, Ferro JM, Tsai FY; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011 Apr;42(4):1158-92. doi: 10.1161/STR.0b013e31820a8364. Epub 2011 Feb 3. PMID: 21293023
31. Ferro JM, Canhão P, Stam J, Bousser MG, Barinagarrementeria F; ISCVT Investigators. Prognosis of cerebral vein and dural sinus thrombosis: results of the International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT). Stroke. 2004 Mar;35(3):664-70. doi: 10.1161/01.STR.0000117571.76197.26. Epub 2004 Feb 19. PMID: 14976332.
32. Shapiro HM, Marshall LF. Intracranial pressure responses to PEEP in head-injured patients. J Trauma. 1978 Apr;18(4):254-6. doi: 10.1097/00005373-197804000-00005. PMID: 351206
33. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129(4):1021-9. doi: 10.1016/j.neuroscience.2004.06.046. PMID: 15561417
34. Klatzo I. Pathophysiological aspects of brain edema. Acta Neuropathol. 1987;72(3):236-9. doi: 10.1007/BF00691095. PMID: 3564903
35. Jha SK. Cerebral Edema and its Management. Med J Armed Forces India. 2003;59(4):326-331. doi:10.1016/S0377-1237(03)80147-8
36. Dreier JP, Lemale CL, Kola V, Friedman A, Schoknecht K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology. 2018 May 15;134(Pt B):189-207. doi: 10.1016/j.neuropharm.2017.09.027. Epub 2017 Sep 21. PMID: 28941738
37. Wendell LC, Khan A, Raser J, Lang SS, Malhotra N, Kofke WA, LeRoux P, Park S, Levine JM. Successful management of refractory intracranial hypertension from acute hyperammonemic encephalopathy in a woman with ornithine transcarbamylase deficiency. Neurocrit Care. 2010 Aug;13(1):113-7. doi: 10.1007/s12028-010-9361-2. PMID: 20422467
38. Bothwell SW, Janigro D, Patabendige A. Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS. 2019 Apr 10;16(1):9. doi: 10.1186/s12987-019-0129-6. PMID: 30967147; PMCID: PMC6456952
39. Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018 Nov;17(11):1016-1024. doi: 10.1016/S1474-4422(18)30318-1. PMID: 30353860; PMCID: PMC6261373
40. Orešković D, Klarica M. Development of hydrocephalus and classical hypothesis of cerebrospinal fluid hydrodynamics: facts and illusions. Prog Neurobiol. 2011 Aug;94(3):238-58. doi: 10.1016/j.pneurobio.2011.05.005. Epub 2011 May 27. PMID: 21641963
41. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004 Jun;16(1):1-13. doi: 10.1016/j.nbd.2003.12.016. PMID: 15207256.
42. Erickson MA, Wilson ML, Banks WA. In vitro modeling of blood-brain barrier and interface functions in neuroimmune communication. Fluids Barriers CNS. 2020 Mar 30;17(1):26. doi: 10.1186/s12987-020-00187-3. PMID: 32228633; PMCID: PMC7106666
43. Erickson MA, Dohi K, Banks WA. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation. 2012;19(2):121-30. doi: 10.1159/000330247. Epub 2012 Jan 11. PMID: 22248728; PMCID: PMC3707010
44. Gadani SP, Walsh JT, Lukens JR, Kipnis J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron. 2015 Jul 1;87(1):47-62. doi: 10.1016/j.neuron.2015.05.019. PMID: 26139369; PMCID: PMC4491143
45. Banks WA. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav Immun. 2015;44:1-8. doi:10.1016/j.bbi.2014.08.007
46. Erickson MA, Banks WA. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol Rev. 2018 Apr;70(2):278-314. doi: 10.1124/pr.117.014647. PMID: 29496890; PMCID: PMC5833009.
47. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005 Jun;57(2):173-85. doi: 10.1124/pr.57.2.4. PMID: 15914466
48. Shi J, Dong B, Mao Y, Guan W, Cao J, Zhu R, Wang S. Review: Traumatic brain injury and hyperglycemia, a potentially modifiable risk factor. Oncotarget. 2016 Oct 25;7(43):71052-71061. doi: 10.18632/oncotarget.11958. PMID: 27626493; PMCID: PMC5342608
49. Durward QJ, Amacher AL, Del Maestro RF, Sibbald WJ. Cerebral and cardiovascular responses to changes in head elevation in patients with intracranial hypertension. J Neurosurg. 1983 Dec;59(6):938-44. doi: 10.3171/jns.1983.59.6.0938. PMID: 6631516
50. Rossi S, Zanier ER, Mauri I, Columbo A, Stocchetti N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. 2001 Oct;71(4):448-54. doi: 10.1136/jnnp.71.4.448. PMID: 11561026; PMCID: PMC1763520
51. Kelly DF, Goodale DB, Williams J, Herr DL, Chappell ET, Rosner MJ, Jacobson J, Levy ML, Croce MA, Maniker AH, Fulda GJ, Lovett JV, Mohan O, Narayan RK. Propofol in the treatment of moderate and severe head injury: a randomized, prospective double-blinded pilot trial. J Neurosurg. 1999 Jun;90(6):1042-52. doi: 10.3171/jns.1999.90.6.1042. PMID: 10350250
52. Kinoshita, K. (2019). Neurocritical Care. In Neurocritical Care. https://doi.org/10.1007/978-981-13-7272-8
Gregory W J Hawryluk, MD, PhD, FRCSC, Andres M Rubiano, MD, Annette M Totten, PhD, Cindy O’Reilly, BS, Jamie S Ullman, MD, Susan L Bratton, MD, Randall Chesnut, MD, Odette A Harris, MD, MPH, Niranjan Kissoon, MD, Lori Shutter, MD, Robert C Tasker, MBBS, MD, Monica S Vavilala, MD, Jack Wilberger, MD, David W Wright, MD, Angela Lumba-Brown, MD, Jamshid Ghajar, MD, PhD, Guidelines for the Management of Severe Traumatic Brain Injury: 2020 Update of the Decompressive Craniectomy Recommendations, Neurosurgery, Volume 87, Issue 3, September 2020, Pages 427–434, https://doi.org/10.1093/neuros/nyaa278
53. Coles JP, Minhas PS, Fryer TD, Smielewski P, Aigbirihio F, Donovan T, Downey SP, Williams G, Chatfield D, Matthews JC, Gupta AK, Carpenter TA, Clark JC, Pickard JD, Menon DK. Effect of hyperventilation on cerebral blood flow in traumatic head injury: clinical relevance and monitoring correlates. Crit Care Med. 2002 Sep;30(9):1950-9. doi: 10.1097/00003246-200209000-00002. PMID: 12352026
54. Laffey JG, Kavanagh BP. Hypocapnia. N Engl J Med. 2002 Jul 4;347(1):43-53. doi: 10.1056/NEJMra012457. PMID: 12097540
Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, Biller J, Brown M, Demaerschalk BM, Hoh B, Jauch EC, Kidwell CS, Leslie-Mazwi TM, Ovbiagele B, Scott PA, Sheth KN, Southerland AM, Summers DV, Tirschwell DL; American Heart Association Stroke Council. 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2018 Mar;49(3):e46-e110. doi: 10.1161/STR.0000000000000158. Epub 2018 Jan 24. Erratum in: Stroke. 2018 Mar;49(3):e138. Erratum in: Stroke. 2018 Apr 18;: PMID: 29367334
55. Hemphill JC 3rd, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, Fung GL, Goldstein JN, Macdonald RL, Mitchell PH, Scott PA, Selim MH, Woo D; American Heart Association Stroke Council; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2015 Jul;46(7):2032-60. doi: 10.1161/STR.0000000000000069. Epub 2015 May 28. PMID: 26022637
56. Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, Fernandes J, Gogichaishvili T, Golden N, Hartzenberg B, Husain M, Ulloa MI, Jerbi Z, Khamis H, Komolafe E, Laloë V, Lomas G, Ludwig S, Mazairac G, Muñoz Sanchez Mde L, Nasi L, Olldashi F, Plunkett P, Roberts I, Sandercock P, Shakur H, Soler C, Stocker R, Svoboda P, Trenkler S, Venkataramana NK, Wasserberg J, Yates D, Yuttha Kasem Unt S; CRASH trial collaborators. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005 Jun 4-10;365(9475):1957-9. doi: 10.1016/S0140-6736(05)66552-X. PMID: 15936423.
57. Rossi S, Zanier ER, Mauri I, Columbo A, Stocchetti N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. 2001;71(4):448-454. doi:10.1136/jnnp.71.4.448
58. Kelly DF, Goodale DB, Williams J, Herr DL, Chappell ET, Rosner MJ, Jacobson J, Levy ML, Croce MA, Maniker AH, Fulda GJ, Lovett JV, Mohan O, Narayan RK. Propofol in the treatment of moderate and severe head injury: a randomized, prospective double-blinded pilot trial. J Neurosurg. 1999 Jun;90(6):1042-52. doi: 10.3171/jns.1999.90.6.1042. PMID: 10350250

Add comment