

14 September 2024, by Shahriar Lahouti.
CONTENTS
- Preface
- Pathophysiology
- Clinical findings
- Laboratory findings
- Differential diagnosis
- Diagnostic evaluation
- Treatment
- Going further
- References
Preface
Toxic alcohols (e.g. ethylene glycol, methanol, isopropyl alcohol) can be found in several settings including hospitals, hardware stores, and the household. Toxic alcohol poisoning presents with various degrees of inebriation, acidemia, and end-organ damage depending on the substance. Timely diagnosis is critical to prevent irreversible organ damage or death and is based primarily on the clinical history and consideration of this entity. Treatment depends on the ingestion and severity of the illness but includes alcohol dehydrogenase blockade with fomepizole or ethanol and special considerations for the initiation of hemodialysis. An understanding of toxic alcohol ingestion can assist emergency clinicians in diagnosing and managing this potentially deadly disease.
Pathophysiology
- Alcohols are rapidly absorbed from the gastrointestinal tract. Once absorbed, they have a volume of distribution similar to that of body water (~0.6 L/kg) with peak blood concentrations occurring within one hour. Subsequently, they are metabolized in the liver or excreted primarily by the kidney.
- The toxic alcohols are inebriating but are not directly toxic, except for isopropanol. Their toxic effects result from their metabolites.
- Metabolic conversion explains biphasic clinical and laboratory findings:
- Initially, clinical findings are due to parent alcohol; later on, they are due to toxic metabolites.
- Laboratory findings initially show elevated levels of parent toxic alcohol; later this disappears, and only acid metabolites are seen.
- Metabolic conversion explains biphasic clinical and laboratory findings:
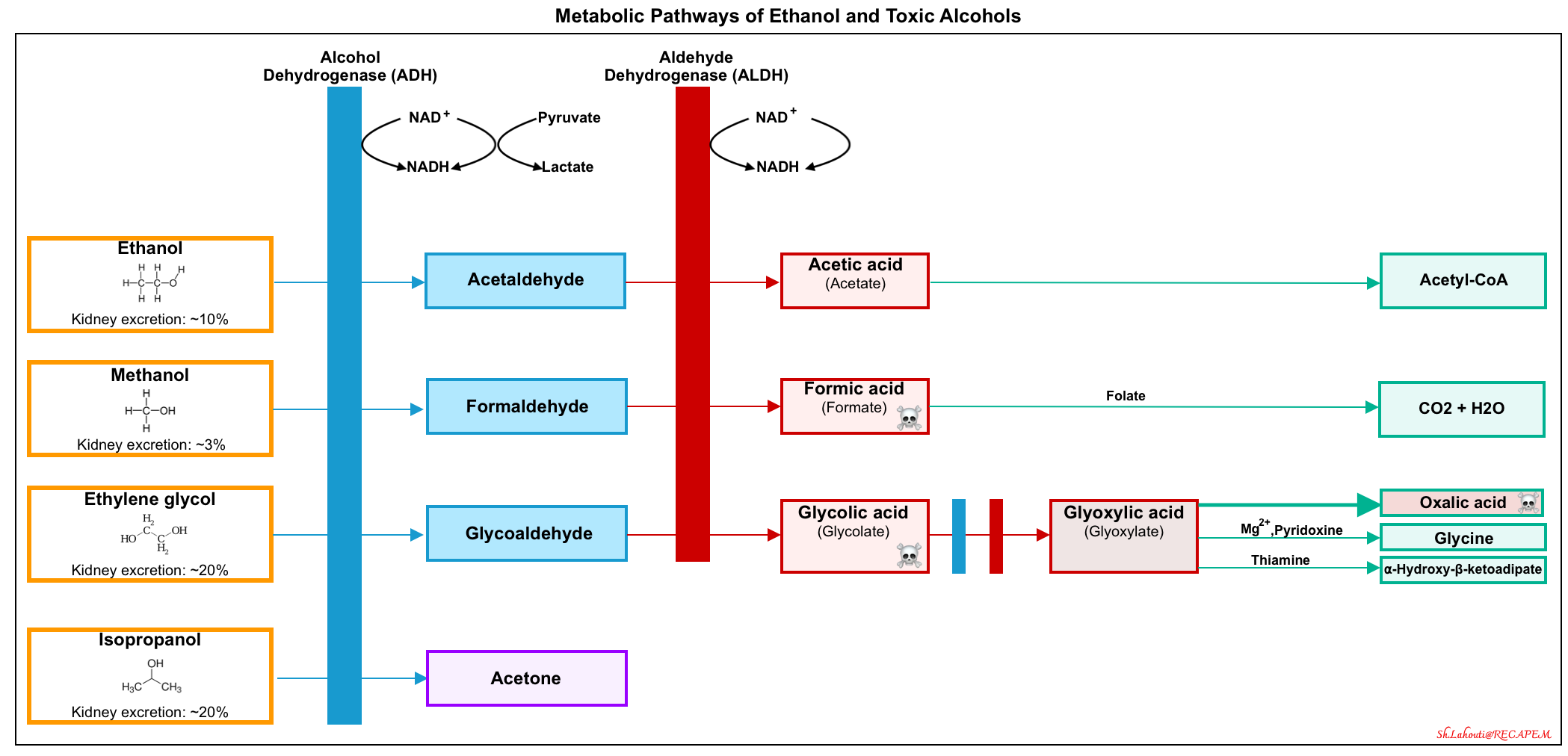
A simplified schema depicting their primary metabolic pathways is shown in the figure below.
| Methanol |
▪️Source:
- Cleaning agents, copy machine fluids, wiper fluid, paint thinners, shellacs, varnishes, deicing fluid, canned heating sources, automotive
fuels, and illegal spirits (moonshine, home-distilled alcohol, etc.)
▪️Metabolism
- Methanol is rapidly absorbed and metabolized in the liver by alcohol dehydrogenase (ADH) to formaldehyde, and then by aldehyde dehydrogenase (ALDH) to formic acid.
- The formate metabolite is then bound by tetrahydrofolate and undergoes metabolism (by 10-formyltetrahydrofolate dehydrogenase) to CO2 and water.
☠️Toxic metabolite: Formic acid
- Formic acid accounts for the toxicity and metabolic acidosis. Formate is a mitochondrial toxin, inhibiting cytochrome oxidase and thereby interfering with oxidative phosphorylation.
- Metabolic acidosis
- Formic acid itself accounts for the metabolic acidosis early in the course of methanol poisoning.
- Lactic acidosis
- Formic acid binds to cytochrome oxidase and blocks oxidative phosphorylation in mitochondria, leading to anaerobic metabolism and the development of lactic acidosis.
- Metabolism of methanol increases the NADH/NAD+ ratio, which favors the conversion of pyruvate to lactate and thereby worsens lactic acidosis *.
- End-Organ toxicity
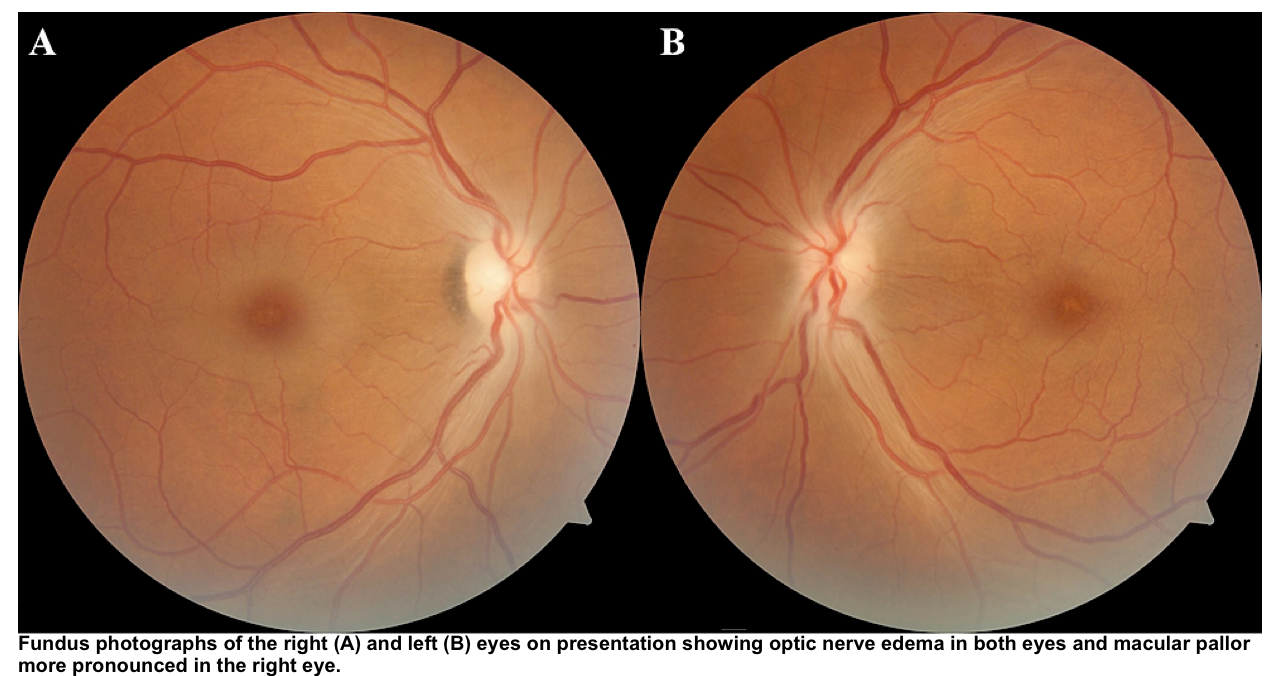
- Ocular toxicity: Retinal pigmented epithelial cells and optic nerve cells appear uniquely susceptible.
- Symptoms:
- Exam findings *:
- Central scotoma
- Both hyperemia and pallor of the optic disc
- Papilledema
- Afferent papillary defect
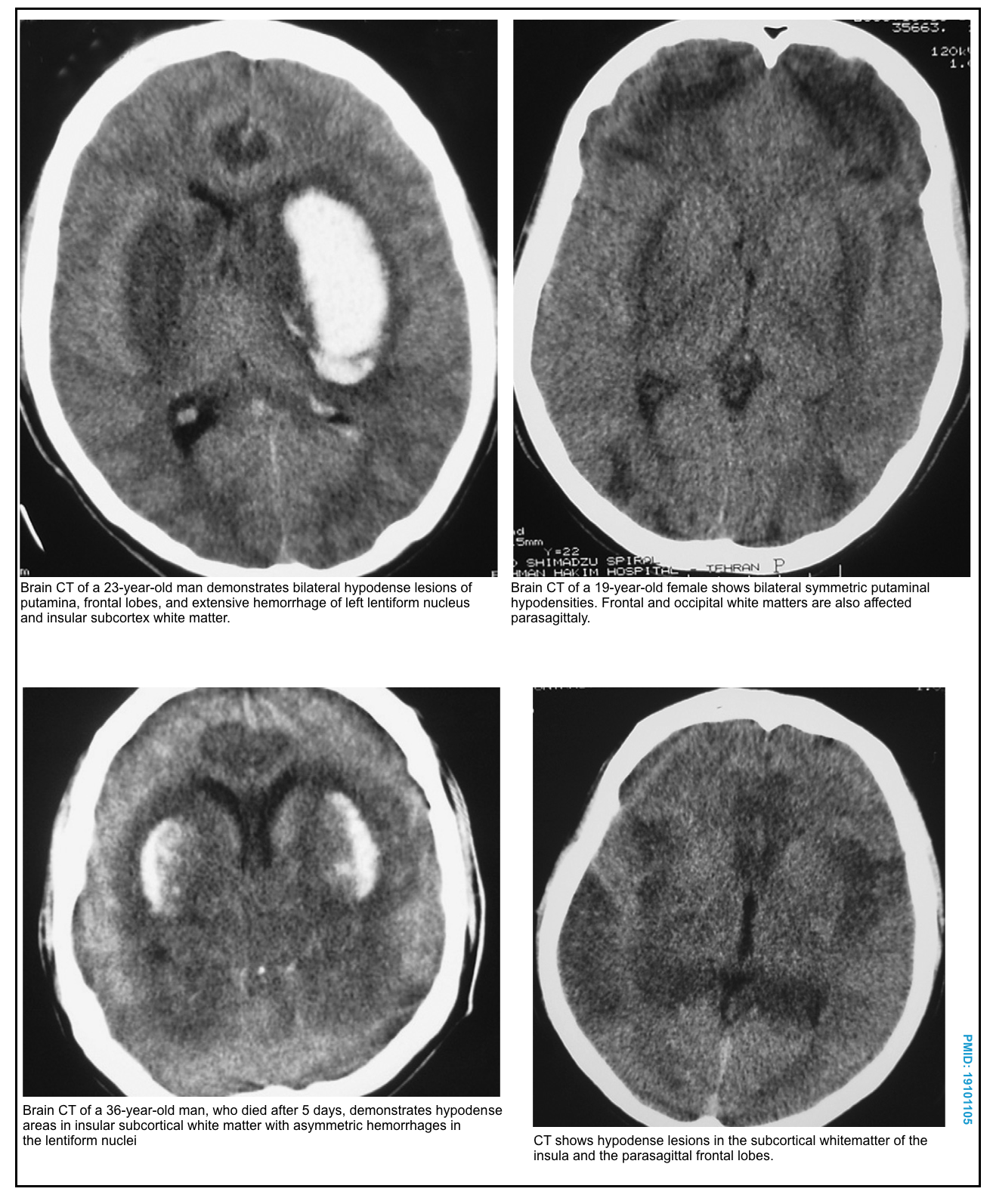
- Basal ganglia neurons
- Signs and symptoms: Parkinsonism * *,*.
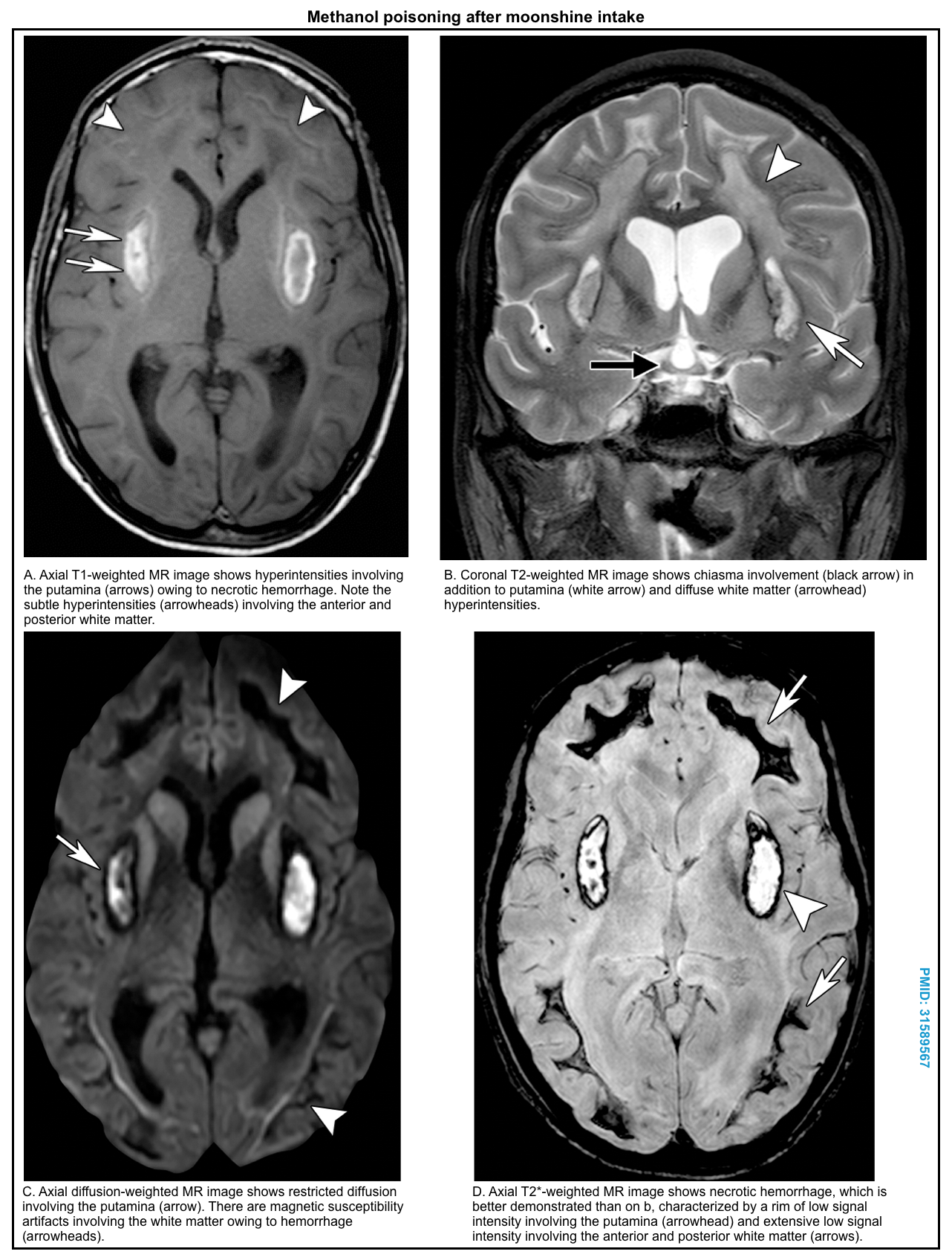
- Bilateral necrosis of the putamen (with or without hemorrhage), and less commonly, caudate nucleus *,*,*.
- Hemorrhagic necrosis appears bright on CT imaging.
- MRI shows hyperintensity on T2/FLAIR sequences, with restricted diffusion. Hemorrhage may be especially notable on SWI/GRE sequences.
- Other CNS lesions include necrosis of the corpus callosum, infarcts of other areas of the brain, and intracranial hemorrhage *,*.
- Ocular toxicity: Retinal pigmented epithelial cells and optic nerve cells appear uniquely susceptible.
- Metabolic acidosis
| Ethylene glycol |
▪️Source
- Detergents, paints, cosmetics, coolant solutions, antifreeze, deicing agents, and hydraulic brake fluid.
▪️Metabolism
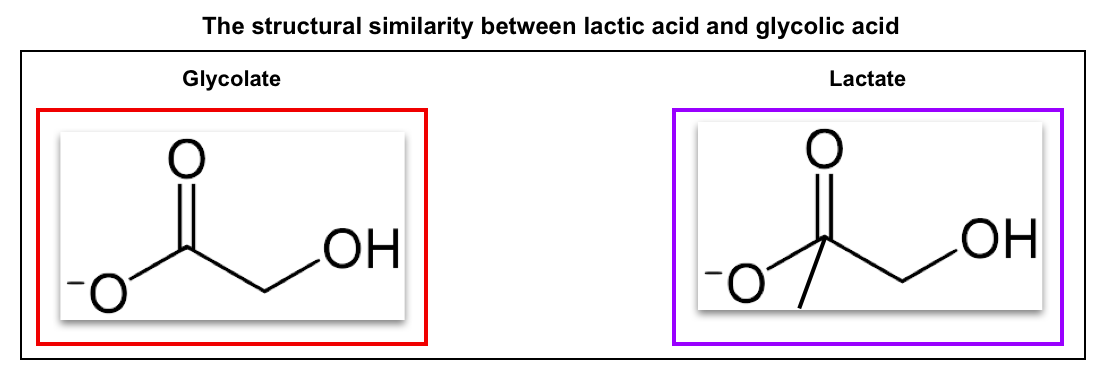
- Ethylene glycol is metabolized by alcohol dehydrogenase to glycoaldehyde, followed by metabolism with aldehyde dehydrogenase to glycolic acid. Subsequently, glycolic acid is converted to glyoxylic acid.
- Glyoxylic acid can be metabolized through several pathways (above figure).
- The major pathway is the conversion of glyoxylic acid to oxalic acid.
- Oxalic acid can be complex with calcium, which leads to hypocalcemia and precipitation of calcium oxalate crystals in tissues and urine.
- End-organ damage from
☠️Toxic metabolite: Glycolic acid and oxalate
- The toxicity of ethylene glycol poisoning is thought to be due to the direct cytotoxicity of glycolic acid and tissue damage from the precipitation of calcium oxalate crystals.
- Metabolic acidosis
- Glycolic acid itself accounts for metabolic acidosis early in the course of ethylene glycol poisoning *.
- Glycolate also impairs cellular respiration contributing to the development of lactic acidosis in some patients *.
- Metabolism of ethylene glycol increases the NADH/NAD+ ratio, which favors the conversion of pyruvate to lactate and thereby worsens lactic acidosis *.
- End-organ damage results from the deposition of calcium oxalate in tissues *
- Nephrotoxicity and AKI *
- Myocardial dysfunction
- Neurologic function
- Pulmonary dysfunction
- Hypocalcemia: Oxalic acid can cause hypocalcemia by precipitation as calcium oxalate, resulting in
- Prolongation of the QT interval on ECG and ventricular dysrhythmias.
- Metabolic acidosis

| Isopropanol |
▪️Source
- Rubbing alcohol, hand sanitizer, and some household cleaners.

▪️Metabolism
- Isopropanol is metabolized by alcohol dehydrogenase to acetone.
- Because acetone is a ketone, not an aldehyde, it cannot be further metabolized by ALDH. It is eliminated in the urine and expired air.
- Acetone does not cause eye, kidney, cardiac, or metabolic toxicity, although high levels of acetone may contribute to CNS depression.
- ⭐️Acetone is a neutral ketone body. It does not cause elevated anion gap or metabolic acidosis.
☠️Toxicity: Isopropanol
Clinical findings
The alcohols initially depress the sensorium and later produce organ dysfunction. Patients may present with symptoms ranging from undifferentiated altered mental status to GI symptoms. The following are the typical symptoms when these are ingested alone. When co-ingested with ethanol, the onset of toxicity may be delayed.
| Ethylene glycol |
▪️Stage 1 (30 min-12 hours): Neurologic stage 🧠
- The clinical findings are mediated by the parent compound “ethylene glycol” and resemble ethanol intoxication *.
- Gastric irritation (pain, nausea, vomiting).
- Inebriation (dizziness, ataxia, confusion, nystagmus).
- May see CNS depression, cerebral edema, and seizure.
▪️Stage 2 (12-24h): Cardiopulmonary stage
- Myocardial dysfunction, shock *.
- Tachypnea, ARDS.
▪️Stage 3 (24-72 hours): Renal stage
- Renal failure due to calcium oxalate crystal deposition in the proximal tubules.
▪️Stage 4 (5-20 days)
- Late neurologic sequelae, e.g. cranial neuropathies can occur.
| Methanol |
▪️Stage 1 (0-6 hours) Neurologic stage 🧠
- The clinical findings are mediated by the parent compound “methanol” and resemble ethanol intoxication *.
- Gastric irritation (pain, nausea, vomiting).
- Inebriation (dizziness, ataxia, confusion, nystagmus).
- Methanol is only a mild inebriant, and patients with tolerance to ethanol also demonstrate tolerance to methanol’s intoxicating effects.
▪️Stage 2 (6-30 hours): A latent phase may occur depending in part on the methanol dose *
- Inebriation resolves.
- It can be asymptomatic.
- ⭐️Consequently, the absence of symptoms and the presence of a clear sensorium do not exclude serious methanol poisoning.
▪️Stage 3 (6-72 hours)
- Visual symptoms (photophobia or blurred or “snow field” vision, and blindness)👁️
- The exam may reveal papilledema, and nonreactive mydriasis once permanent damage has occurred *.
- Seizure, coma, cerebral edema, herniation.
- Cardiac failure and respiratory arrest can occur.
| Isopropanol |
- The onset of symptoms occurs within 30 to 60 minutes after ingestion.
- GI symptoms are prominent, and range from nausea, vomiting, abdominal pain, and acute pancreatitis to hemorrhagic gastritis and upper GI bleeding *.
- Inebriation (dizziness, ataxia, confusion, nystagmus).
- May see CNS depression, cerebral edema, and seizure.
- Cardiovascular: Hypotension; secondary to peripheral vasodilation *.
Laboratory findings
Metabolic acidosis
- Metabolic acidosis with an elevated anion gap is a hallmark of toxic alcohol poisoning. This is the consequence of the metabolism of the alcohols to toxic organic acids.
- An exception is isopropanol which is metabolized to acetone.
- Acetone is a neutral ketone. Ketosis without acidosis is essentially diagnostic of isopropanol poisoning.
- An exception is isopropanol which is metabolized to acetone.
Anion Gap Elevation
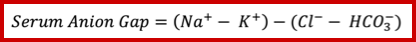
▪️Background
- A normal anion gap is roughly 4-12 mM.
- Historically, the normal range of anion gap was often quoted as being higher (e.g. up to ~16 mM). However, with newer electrolyte analyzers, the upper limit of normal has decreased to ~11-12 mM *.
- Comparison with a baseline anion gap is helpful if that is available. For example:
- Patients with chronic renal insufficiency may often have a chronically elevated anion gap. If their anion gap is at its chronic level, this is reassuring.
- If the anion gap is rapidly elevating over time, this is worrisome and requires aggressive investigation.
- Remember that the elevated anion gap is not specific to toxic alcohol ingestion (see below figure).
- Of note, acetone is a neutral ketone body and does not cause acidosis or elevated anion gap. In contrast, the two other ketone bodies (acetoacetate and Beta-hydroxybutyrate) are acidic and cause ketoacidosis with a high anion gap *.
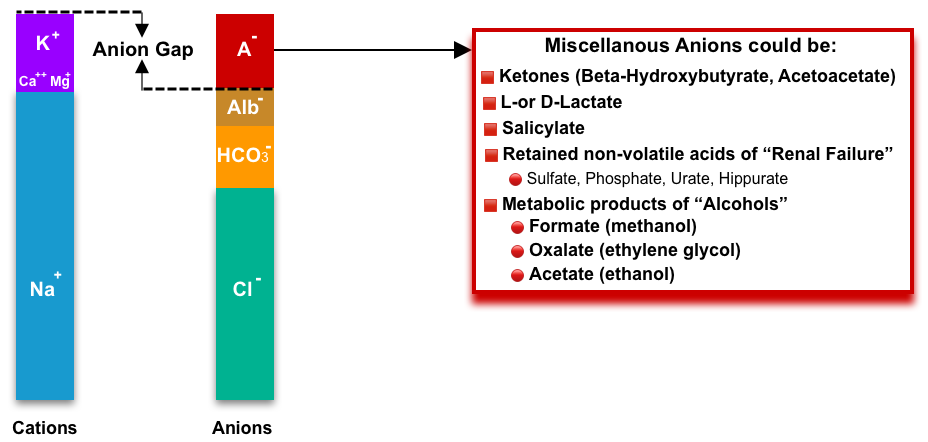
🔴Ketoacidosis. More on this, here.
🔴Uremic acidosis
🔴Elevated L-lactate
🔴Salicylate ingestion. More on this, here.
🔴Metabolic product of toxic alcohols
🔴Hyperphosphatemia
🔴Metabolic alkalosis (↑the negative charge on albumin).
🔴D-hyperlactatemia (Due to small intestinal resection or malabsorption that causes carbohydrates to be delivered to the colon and fermented by gut bacteria. Note that laboratory tests for “lactate” will not detect D-lactate. D-hyperlactatemia may cause neurological symptoms including confusion, ataxia, incontinence, and nystagmus.)
▪️Anion gap in toxic alcohol poisoning
- The cause of the elevated anion gap in toxic alcohol poisoning is primarily the organic acid metabolites.
- The relation between the time course of changes in the osmolar gap and anion gap in toxic alcohol ingestion is shown below.
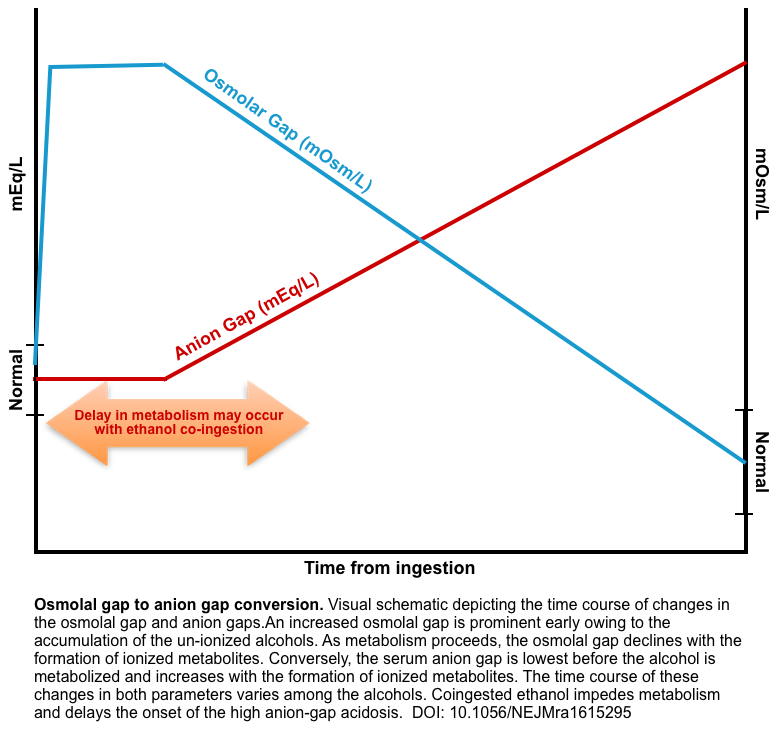
- Does a normal anion gap rule out toxic alcohol ingestion?
- We already knew that clinically significant poisoning with ethylene glycol or methanol will cause anion gap elevation (often extremely high). However, the generation of organic acids may be delayed for several hours (explained above).
- Co-ingestion of ethanol can further delay toxic organic acid formation since alcohol dehydrogenase (ADH) has a stronger affinity for ethanol compared to toxic alcohols. Of note, if a patient has a blood ethanol level of > 70 mg/dL, they are unable to metabolize the toxic alcohol to the organic acid necessary to generate an anion gap in metabolic academia *.
- Therefore, if a patient has an ethanol level of > 70mg/dL, to have an anion gap metabolic acidemia secondary to a toxic alcohol, one should wait 2-12 hours for toxic alcohol to be metabolized.
- The issue is when we can expect to see this elevation in the anion gap.
- Based on the current evidence, serial measurement of anion gap over time should be extremely sensitive to toxic alcohol poisoning. This might be accomplished as follows:
- Measure the anion gap at baseline and then repeat every 2 to 4 hours.
- The duration of the following anion gap is unclear.
- For ethylene glycol without ethanol co-ingestion, 4-6 hours of following the anion gap is enough.
- For patients with ethanol co-ingestion, the anion gap should be cycled until 6 hours after the ethanol is metabolized.
- A recent review suggested cycling the anion gap for 12 hours *.
- The intensity and duration of anion gap measurement could be adjusted based on clinical scenarios and the index of suspicion.
- For example, if the patient had a toxic ingestion >6 hours previously and ethanol levels are negative, then serial anion gap measurements might not be needed at all.
- Based on the current evidence, serial measurement of anion gap over time should be extremely sensitive to toxic alcohol poisoning. This might be accomplished as follows:
- We already knew that clinically significant poisoning with ethylene glycol or methanol will cause anion gap elevation (often extremely high). However, the generation of organic acids may be delayed for several hours (explained above).
Lactate & Lactate Gap
▪️Background
- Lactic acid is an organic acid. It has two enantiomers: L-lactic acid and D-lactic acid.
- L-lactate composes nearly the whole of lactate present in human beings because mammalian cells exclusively contain L-lactate dehydrogenase (LDH); the enzyme that converts pyruvate to lactate (PYR→ Lac).
- In the normal physiologic state, the blood concentration is 0.5-1 mmol/L.
- D-lactate is produced by bacteria. In human beings, this is produced in nanomolar concentrations by GI bacterial flora *. However, it may accumulate in certain pathologic conditions and cause metabolic acidosis. Of note, D-lactic acidosis is not easily identified because D-lactate and L-lactate have the same physical and chemical properties, and the normal analytical methods are not able to distinguish the two isomers *.
- L-lactate composes nearly the whole of lactate present in human beings because mammalian cells exclusively contain L-lactate dehydrogenase (LDH); the enzyme that converts pyruvate to lactate (PYR→ Lac).
- In the following discussion, L-lactate is referred to as lactate unless otherwise specified.
- Production of lactic acid
- Source: The main sources of intracellular lactate are glucose (~65%) and alanine (~25%) through their conversion into pyruvate (below figure) *.
- Lactate formation from glucose: In the last step of glycolysis (during normal metabolism), pyruvate is converted to lactate in the cytoplasm by LDH while NADH is oxidized to NAD+ *.
- Lactate formation from alanine: The enzyme alanine aminotransferase (ALT), catalyzes the conversion of alanine to produce pyruvate in the
cytoplasm. Pyruvate is then reduced to lactate by LDH *.
- ⭐️This cytosolic regeneration of NAD+ by LDH is mandatory for glycolysis to continue, as NAD+ is needed for the glycolytic reaction that converts glyceraldehyde 3-phosphate into 1,3-bisphosphoglycerate. Without an efficient mechanism in the cytosol to recycle NAD+ from NADH, glycolysis cannot happen.
- Source: The main sources of intracellular lactate are glucose (~65%) and alanine (~25%) through their conversion into pyruvate (below figure) *.
- Removal of lactic acid
- The lactate is removed principally via two mechanisms
- Gluconeogenesis (production of glucose from lactate)
- Liver (~60%).
- Renal cortex (~30%)
- Liver (~60%).
- Oxidation as a fuel (10%). This happens in many organs (liver, kidney, muscle, heart, and brain) *.
- Gluconeogenesis (production of glucose from lactate)
- The lactate is removed principally via two mechanisms
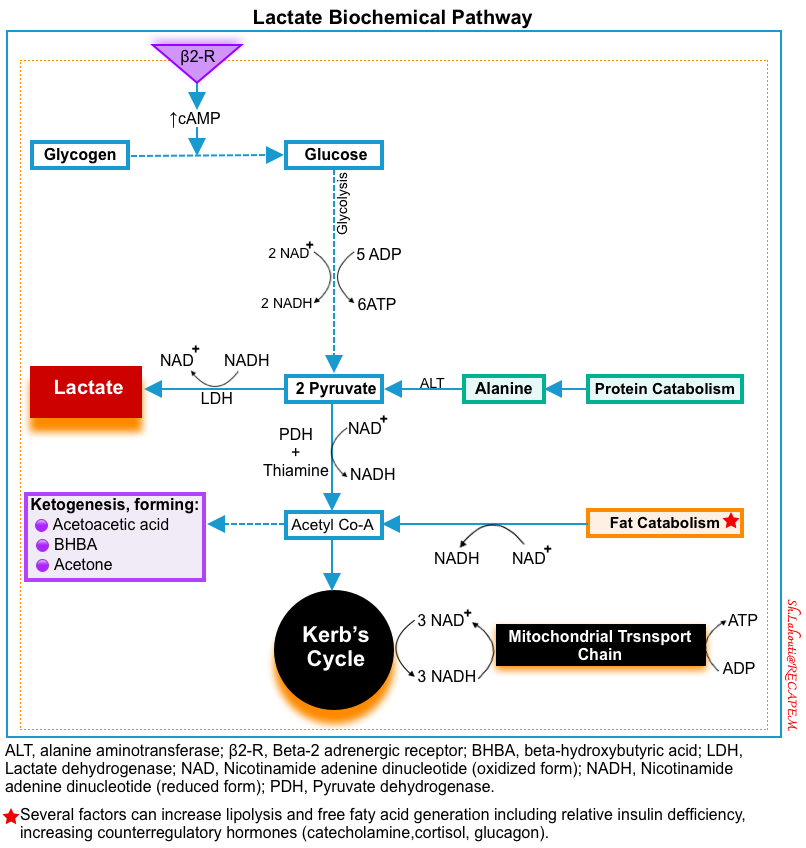
▪️Causes of hyperlactatemia
- Increased lactate production
- Increased glycolysis and pyruvate production
- IV glucose administration.
- Beta-2 agonist activity (stimulates glycogenolysis and increases the concentration of pyruvate) *.
- Increase in NADH/NAD+ ratio (i.e. NAD+ deficiency)
- Mitochondrial dysfunction due to hypoxemia.
- Mitochondrial dysfunction for any other reason (e.g. medications).
- Low insulin levels or insulin resistance (e.g. ketoacidosis states).
- Increased glycolysis and pyruvate production
- Decreased lactate clearance
- Several factors are associated with decreased hepatic clearance, including acidosis, underlying cirrhosis, and hypoperfusion *.
🔴Type A: generalized or regional tissue hypoxia
-Shock of any etiology
-Regional hypoperfusion: Mesenteric ischemia, ischemic limbs
-Muscle hyperactivity, e.g. generalized seizure, extreme exertion.
-Extreme anemia (e.g. <4 g/dL).
-Systemic hypoxemia
-CO poisoning
🔴Type B1: systemic diseases
-Liver failure.
-Thiamine deficiency
🔴Type B2: drugs
-Beta-agonist excess, e.g. epinephrine, albuterol.
-Linezolid.
-Metformin: Virtually, metformin is a mitochondrial toxin, which inhibits the mitochondrial transport chain-I
🔴Type B2: toxins
-Alcohols: Ethylene glycol, Methanol, Ethanol poisoning (usually <5 mM).
-Acetaminophen poisoning (massive)
-Carbon monoxide.
-Cyanide.
-Salicylate.
-Sympathomimetics (cocaine, amphetamine, cathinone).
-Toluene.
-Iron.
🔴Type B3: inherited
-MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes).
🔴Type D: D-Lactate Excessive production
-Short Bowel Syndrome
-Jejunoileal bypass surgery
-In a situation of abdominal compartment syndrome
-Poisoning from heavy metals
-Some infections and sepsis *
🔴Elevated lactate in alcohol poisoning
- Patients with toxic alcohol poisoning can have a true elevation of their lactate levels, for a variety of reasons:
- Methanol results in formic acid production, which is a mitochondrial poison.
- Glycolate also impairs cellular respiration contributing to the development of lactic acidosis in some patients *.
- Metabolism of toxic alcohols increases the NADH/NAD+ ratio, which favors the conversion of pyruvate to lactate and thereby worsens lactic acidosis.
- Patients are often alcoholics, with low thiamine levels.
- Generally, the level of lactate is fairly low (e.g. <5 mM).
- ⭐️The degree of lactate elevation is generally too low to fully account for the elevated anion gap (anion gap elevation is due primarily to other anions, such as formate and glyoxylate).
🔴Lactate gap in ethylene glycol poisoning
- In general, lactate measurement is based on two different methods:
- LDH (lactate dehydrogenase)-based measurement of lactate
- This is accurate.
- Generally utilized by laboratory assays.
- Lactate oxidase-based method
- This assay is often utilized in point-of-care devices.
- This method can be falsely elevated in the presence of glycolate *.
- LDH (lactate dehydrogenase)-based measurement of lactate
- If a lactate gap is detected (a deviation between two measurements), this is suggestive of ethylene glycol poisoning.
Osmolar gap
Accumulation of the alcohol increases the serum osmolality and the osmolal gap (the difference between the serum osmolality measured by the freezing-point depression and the serum osmolarity calculated from the equation given below). The osmolar gap is a surrogate test for the parent alcohol that raises the patient’s serum osmolarity.
Osmolar Gap 🧮 = [measured serum osmolarity] − [calculated serum osmolarity]
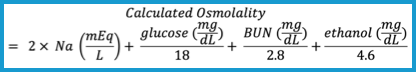
⚠️Osmolar gap pitfalls
- There is wide human variability in the normal values of the osmolar gap, with a range of −10 to +10 *.
-
Patients presenting late may have metabolized most of the parent alcohol, resulting in a low or absent osmolar gap.
-
However, these patients are likely the most sick, requiring resuscitative interventions and hemodialysis.
-
-
Patients who have a negative osmolar gap at baseline could have a seemingly normal osmolar gap, even after toxic alcohol ingestion.
-
Therefore, the osmolar gap is not a definitive rule-out test in high pre-test probability patients.
-
-
Other osmotically active substances and medical conditions can result in a false-positive osmolar gap, including alcoholic ketoacidosis, diabetic ketoacidosis, renal failure, or shock *.
- More on this 👉 Toxicology dogmalysis: the osmolal gap (Josh Farkas)
Differential diagnosis
◾️Toxic alcohol ingestion can be easily confused with many other underlying pathologies.
- Patients may present with symptoms ranging from undifferentiated altered mental status to GI symptoms.
- Acidemia should be present in late presenting cases of toxic alcohol ingestion, except for isopropyl alcohol, though depending on the time of ingestion and co-investments, a significant acidemia may not be appreciated.
- In cases of ethylene glycol that present late in the disease course, clinical clues may include evidence of myocardial depression (cardiogenic shock, reduced cardiac contractility, ECG demonstrating ischemia), cerebral edema (CT scan with effacement of sulci, Cushing triad), metabolic acidemia, and hypocalcemia.
- Late presenting sub-fatal methanol ingestion patients may present with a chief complaint of vision loss.
- Isopropyl alcohol intoxication mainly presents with evidence of inebriation, though some patients may present with signs of gastric irritation (e.g. nausea and vomiting).
😔Altered level of consciousness. More on this, 👉 here.
🤮Nausea & vomiting. See👉 here.
🩸Conditions associated with ↑anion gap metabolic acidosis:
- Ketoacidosis
- Lactic acidosis
- Uremic acidosis
- Salicylates. More on this here.
- Toxic alcohols (methanol, ethylene glycol, diethylene glycol, propylene glycol).
Few conditions can cause a profound high anion gap metabolic acidosis (serum bicarbonate <8 meq/L). The following list includes some causes of single-digit bicarbonate (however it’s not comprehensive) *
🔳Post-cardiac arrest
🔳Profound shock
🔳Liver failure
🔳Mesenteric ischemia
🔳Limb ischemia
🔳Status epilepticus
🔳Severe ketoacidosis: Diabetic ketoacidosis, Alcoholic ketoacidosis
🔳Toxicological etiologies:
-Toxic alcohol ingestion.
-Metformin poisoning.
-Salicylate intoxication.
-Cyanide toxicity.
Diagnostic evaluation
🔴Lab tests to consider when toxic alcohol exposure is considered
- Serum methanol, ethylene glycol, and isopropyl alcohol concentrations.
- Elevated only early in intoxication.
- Levels >20 mg/dL are considered potentially toxic (or methanol >6.2 mM or ethylene glycol >3.2 mM).
- Ethanol level.
- Electrolytes including Ca/Mg/Phos (with anion gap determination). See above.
- Lactate & Beta-hydroxybutyrate levels.
- If the anion gap and/or lactate are elevated: check both a point-of-care lactate and a simultaneous lactate measured in the laboratory to evaluate for a lactate gap.
- Urinalysis
- In cases where ketonuria or ketonemia is detected, isopropyl alcohol ingestion should be considered. In these cases, an elevated beta-hydroxybutyrate may help differentiate between isopropyl alcohol ingestion and alcoholic ketoacidosis or euglycemic diabetic ketoacidosis.
- BUN and creatinine
- Creatinine kinase.
- Serum osmolality.
- Methemoglobin level (if there is cyanosis following the ingestion of antifreeze, which may contain nitrates).
🔵Basic tests: Routine evaluation of any poisoned patient should include the following
- Fingerstick glucose to rule out hypoglycemia as the cause of any alteration in mental status
- Acetaminophen and salicylate levels to rule out these common coingestions
- ECG to rule out conduction system poisoning by drugs that affect the QRS or QTc interval.
- Of note, advanced ethylene glycol poisoning can affect conduction and prolong the QTc interval via its effects on serum calcium *.
- Pregnancy test in women of childbearing age.
- Brain CT/MRI (in patients with altered level of consciousness). Brain MRI may suggest the underlying cause of toxic metabolic encephalopathy (shown below) *.
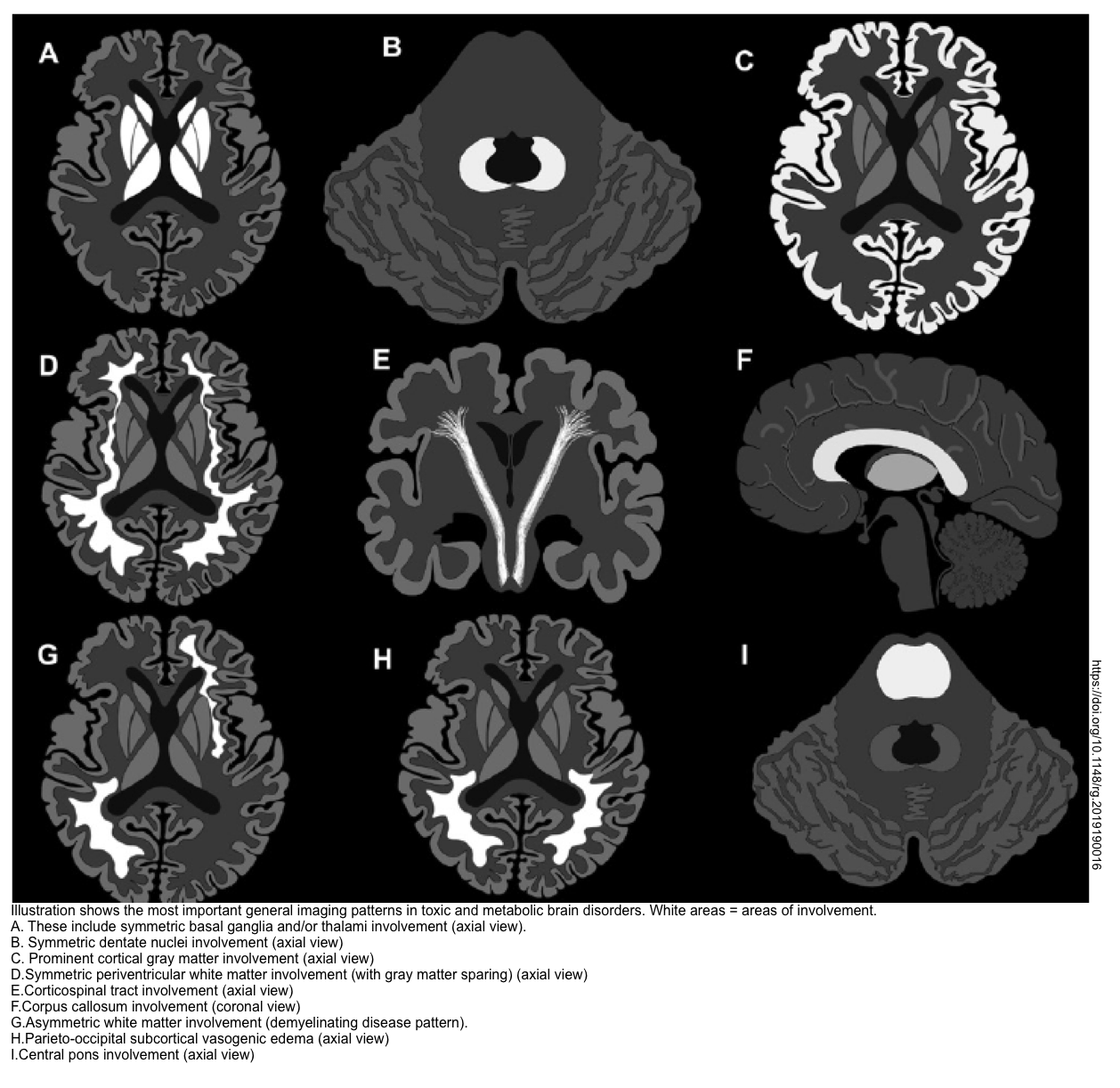
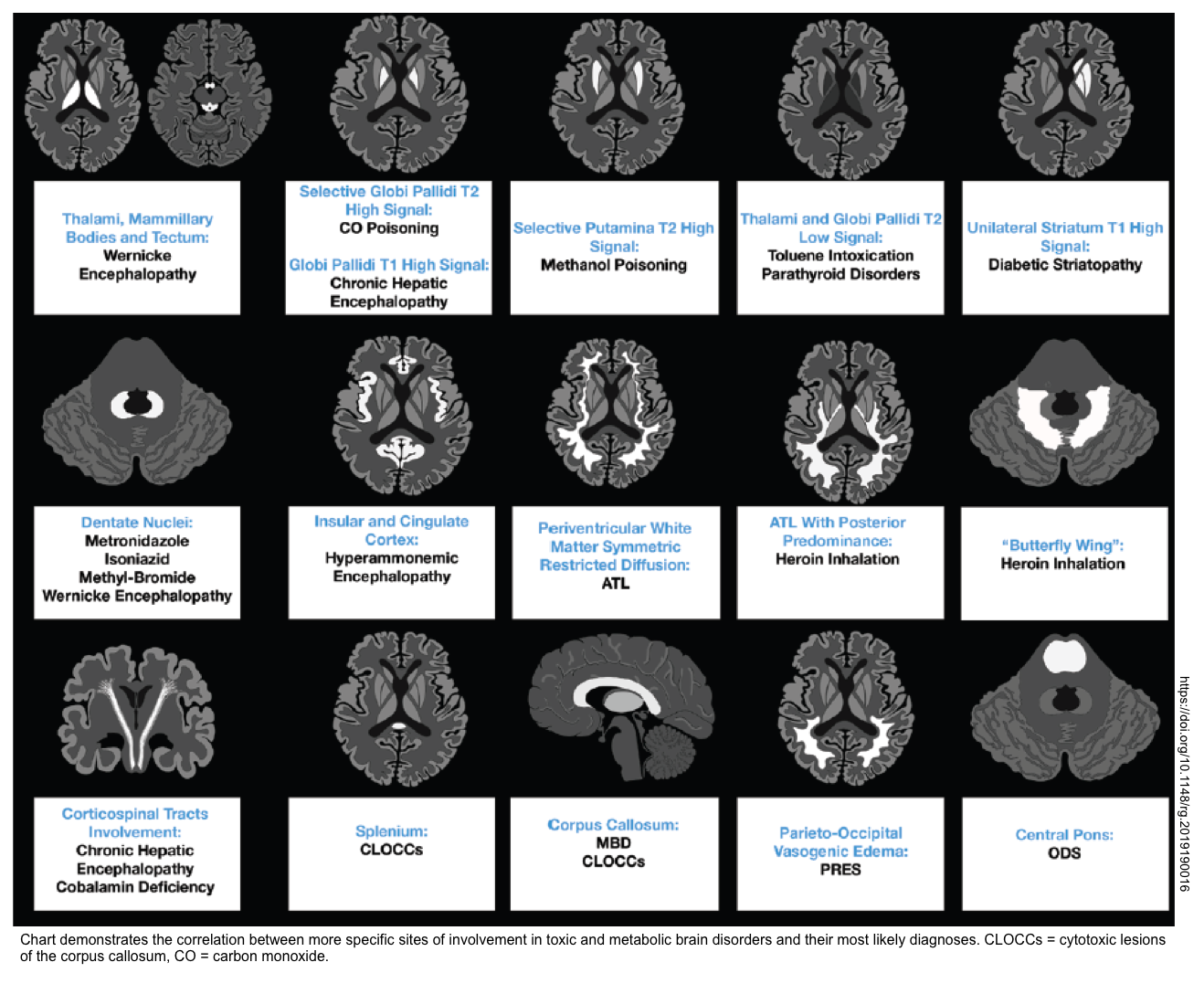
Table summary
- A summary of useful diagnostic findings is presented below.
| Alcohol Type | Minimum lethal dose without treatment | Substance(s) Causing Toxicity | Lab abnormalities | Useful clinical findings |
| Methanol | 1 gram/kg or about 100 mL in an adult | ☠️Formic acid: Mitochondrial toxin. | ↑Osmolal gap HAGMA ↑Formate Lactic acidosis | Gastric irritation & Inebriation Visual symptoms Seizure, coma, cerebral edema |
| Ethylene glycol | 1.1–1.7 grams/kg or about 100 mL in an adult | ☠️Glycolic acid ☠️Calcium oxalate | ↑Osmolal gap HAGMA Calcium oxalate crystalluria ↓Serum Ca Lactate gap* | Gastric irritation & Inebriation & Seizure Myocardial dysfunction, shock Renal failure |
| Isopropanol | 2 to 4 mL/kg up to 1L in adult | ☠️Isopropanol | ↑Osmolal gap Ketonemia Ketonuria Note that HAGMA is absent. | Gastric irritation & Inebriation Hypotension (peripheral vasodilation) |
Treatment
General principles
- Patients presenting early after ingestion may show few signs or symptoms despite potentially life-threatening ingestion and require prompt diagnostic evaluation and treatment.
- As with the treatment of all critically ill patients, airway, breathing, and circulation are the initial treatment priorities.
- Airway, breathing
- If mechanical ventilation is necessary, a higher-than-normal minute ventilation is required given the severe metabolic acidosis.
- Fluid resuscitation should be provided to achieve euvolemia.
- Sodium bicarbonate should be considered for severely acidotic patients, especially for methanol poisoning while awaiting hemodialysis. This is not a substitute for hemodialysis, but a temporizing measure.
- Benzodiazepines for seizures during the initial resuscitation.
- Hypocalcemia can be seen in ethylene glycol poisoning. Avoid giving calcium if possible (this may exacerbate the precipitation of calcium oxalate within tissues).
- Calcium is indicated for tetany, seizures, or substantial QT prolongation *.
- Airway, breathing
- Antiemetics for the treatment of GI distress.
- Treatment mirrors these biphasic metabolism features.
- Early on: fomepizole, to inhibit alcohol dehydrogenase (and thus prevent metabolite generation).
- Later on: once metabolites have been generated, dialysis may be needed to remove them.
- Once there has been confirmed ingestion of toxic alcohol, or if there is high suspicion, the primary treatment priority is to stop the conversion of the toxic alcohol by alcohol dehydrogenase. Obtain early nephrology consultation for acidotic patients, even if confirmatory levels are not yet obtained; as these patients may require hemodialysis.
Alcohol dehydrogenase blockade
▪️Indications
-
Significant accidental ingestion of toxic alcohol.
-
Suspicion for ingestion of a toxic alcohol.
-
High anion gap acidosis and/or high osmolar gap (osmolar gap >10) of unknown cause.
-
Serum ethylene glycol or methanol concentration ≥20 mg/dL (ethylene glycol 3.22 mmol/ L or methanol 6.24 mmol/L.
- Generally, waiting for these labs to come back delays therapy, so treatment should be initiated empirically as below.
▪️When to stop alcohol dehydrogenase blockade
- Stop when levels of toxic alcohol are known to be undetectable or at a safe level (<20 mg/dL, or methanol <6.2 mM, or ethylene glycol <3.2 mM).
- Unfortunately, elimination of ethylene glycol or methanol may take a while:
- Ethylene glycol is renally cleared with a half-life of 17 hours (longer in patients with renal insufficiency).
- Methanol is cleared via respiration, with a half-life of ~50 hours.
- In many cases, dialysis may be required to clear parent alcohols and facilitate discharge from the hospital. Alcohol dehydrogenase blockade can be stopped some hours after hemodialysis, once labs confirm that there is no rebound in toxicity.
| Alcohol Dehydrogenase blockade | General dosing | Considerations |
| Fomepizole | 15 mg/kg IV loading dose, followed by 10 mg/kg every 12 hours for 4 doses, followed by 15 mg/kg every 12 hours until ethylene glycol or methanol levels have decreased to < 20 mg/dL | In patients undergoing dialysis, fomepizole should be re-administered every 4 hours |
| Ethanol IV | 10 mL/kg 10% ethanol IV then Maintenance infusion 1 mL/kg/hour for a target serum concentration of 100 mg/dL until ethylene glycol or methanol levels have decreased to < 20 mg/dL | |
| Ethanol PO | Distilled spirits of 40-50% ethanol concentration diluted to 20% and administered as 5 mL/kg loading dose then 0.5 mL/kg/hour maintenance for a target serum concentration of 100 mg/dL -or- *Loading dose of 3-4 standard drinks, followed by a maintenance dose of 1-2 standard drinks per hour with a goal concentration of 100mg/dL | *1 standard drink is 12-14g of ethanol. Examples of standard drinks equivalents: one 12 oz regular beer at 5%, one 5 oz glass of wine at 12%, one 1.5 oz shot of distilled spirits at 40% |
Cofactors
💡Rational: To shunt toxic alcohols toward their non-toxic metabolites.
◾️For ethylene glycol
- Thiamine 100 mg IV daily.
- Pyridoxine (vitamin B6) 100 mg IV twice daily.
◾️For methanol
- Folinic acid (a.k.a. leucovorin), 50-100 mg IV q4hr.
- If unavailable, folic acid may be used at the same dose.
Sodium bicarbonate
◾️Background
- Normalizing pH could keep toxic metabolites in an ionized state (e.g. formate, glycolate). This is beneficial for two reasons:
- Molecules in the ionized state are less able to penetrate tissues (e.g. the brain and retina).
- Molecules in the ionized state pass into the renal tubules, get “trapped” there, and are excreted in urine.
- Stabilizing the pH with bicarbonate is not intended as an alternative to hemodialysis, but rather as a bridge to hemodialysis.
🎯Target pH
- The therapeutic target should probably be a normal pH (this isn’t alkalinizing the blood, but rather just bringing the pH back to a normal level).
◾️Administration
- Bolus: Hypertonic bicarbonate
- 1-2 mEq/kg of hypertonic NaHCO3 IV push to rapidly improve the pH.
- Maintenance infusion: Isotonic bicarbonate
- 3 amps of NaHCO3 8.4% (150 mEq) in 1 L of D5W.
- Infusion rate at 2 × maintenance.
Hemodialysis
🔳Role
- Hemodialysis removes the parent’s toxic alcohols and their metabolites (e.g. formate, oxalate, and glycolate).
- Shorten the duration of fomepizole therapy required, and the hospital length of stay.
- Correct metabolic derangements.
🔳Indications *
- Acidosis
- Continued acidosis ≤ 7.15 despite standard supportive measures.
- Anion gap > 24
- End-organ damage
- Patients with visual field deficits, seizures, or in a state of coma
- Renal failure (this may also inhibit the clearance of various substances).
- Methanol or ethylene glycol level:
- >50 mg/dL in the absence of ethanol or fomepizole therapy.
- >60 mg/dL in the context of ethanol therapy.
- >70 mg/dL in the context of fomepizole therapy.
◾️Considerations
- Intermittent hemodialysis is preferred, to remove toxins rapidly.
- Methanol poisoning may cause coagulopathy, so be careful if anticoagulation is being used to facilitate dialysis.
- Continue dialysis until the alcohol level is <20 mg/dL and acidosis resolves.
- A repeat level may sometimes be obtained two hours after hemodialysis to exclude rebound.
- Continue alcohol dehydrogenase blockade during dialysis.
- Dose adjustment may be required to maintain adequate levels. For example, fomepizole needs to be dosed q4hr.
Going further
- Toxic Alcohols – Minding the Gaps (Anton Helman)
- Toxic Alcohol Ingestion (Life In The Fast Lane)
- TOXIC ALCOHOL POISONING
Add comment