


June 21, 2021 by Shahriar Lahouti. Last updated November 5, 2024.
CONTENTS
- Preface
- Basic physiology
- Definition and diagnosis of DKA
- Severity of DKA
- Differential diagnosis
- Management
- Complications
- Special situations
- RECAP
- Media
- Guidelines
- Appendix
- Going further
- References
Preface
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) are two of the most serious acute complications of diabetes with overlapping features. Despite advances in monitoring technologies, insulin therapeutics, and insulin delivery systems, the rate of hyperglycemic emergencies remains largely unchanged. A better understanding of the pathophysiology and a timely diagnosis, comprehensive clinical and biochemical evaluation, and effective management have been shown to reduce mortality rates.
Basic physiology
The three essential fuels for cellular metabolism are glucose, fatty acids, and amino acids. Insulin and counterregulatory hormones can alter the metabolic milieu of carbohydrate, protein, and lipid metabolism. The prominent metabolic effects of insulin on glucose include *:
- Decreased hepatic glucose production ( ↓glycogenolysis, ↓gluconeogenesis, ↑glycogen synthesis)
- Increased glucose uptake in peripheral tissue e.g. fat, muscles, etc.
In most tissues, glucose is the cardinal cellular fuel. The availability of glucose for intracellular metabolism depends upon adequate serum glucose and sufficient amounts of insulin. When glucose is unavailable, the primary cellular fuel menu flips from glucose to ketone body consumption.
The production of ketone bodies (ketogenesis) takes place in the liver. When carbohydrate availability is significantly reduced, the body resorts to the use of ketones as a primary energy source. A series of biochemical reactions and regulatory hormones tightly regulate this process (figure below).
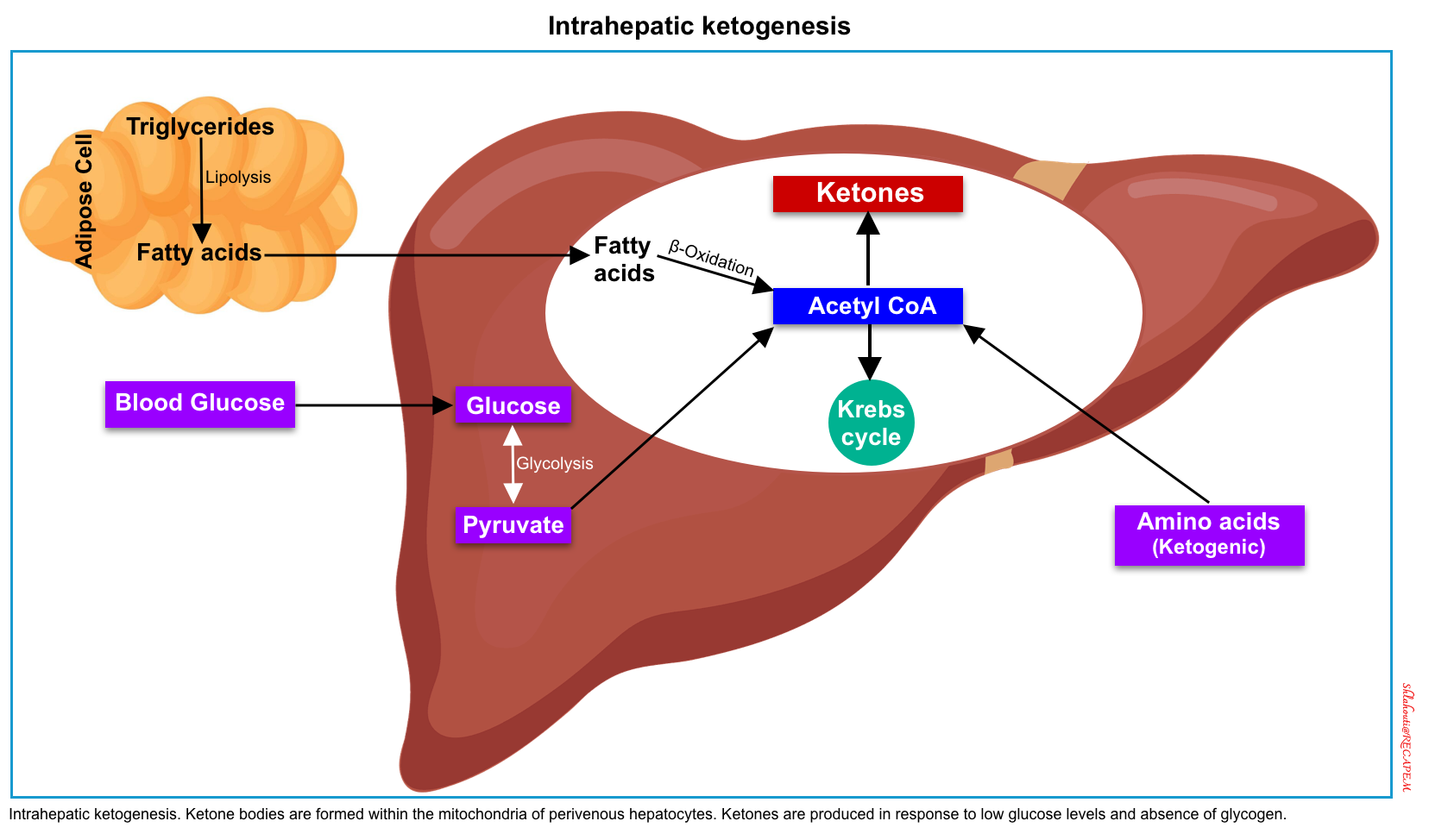
Ketone metabolism is affected by age, the fasting versus fed state, basal metabolic rate, macronutrient intake and balance, liver glycogen stores, and amino acid mobilization from muscle *.
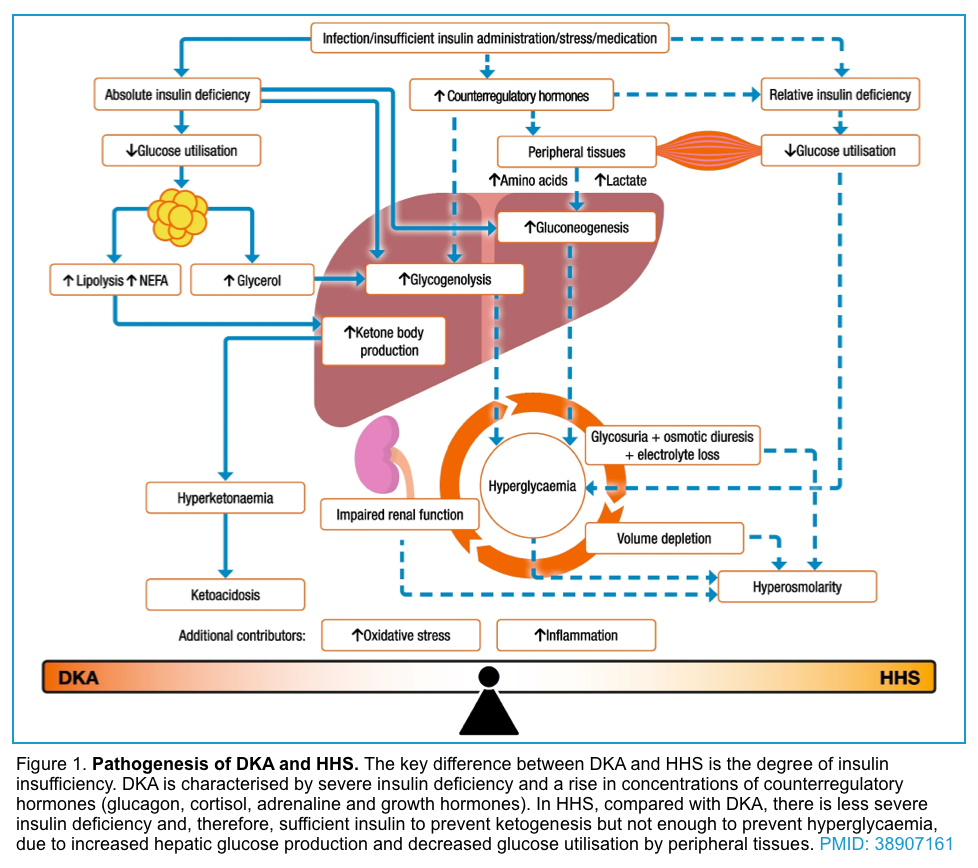
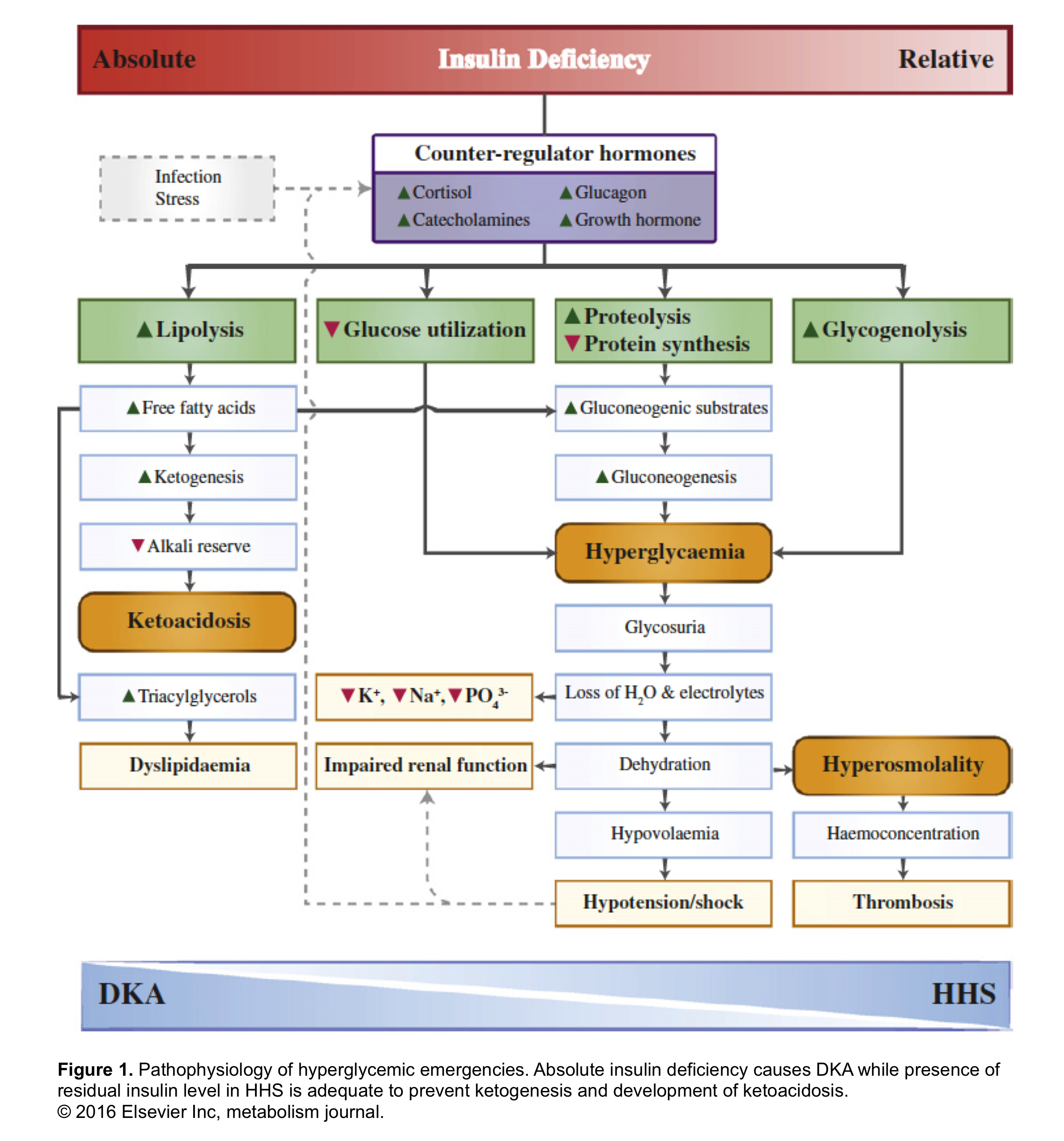
In summary, insulin deficiency and or resistance, glucagon excess, and increased secretion of catecholamines, cortisol, and growth hormone (which oppose the actions of insulin), also contribute to the increases in glucose and ketoacid production (figure 1 below).
- Suppression of ketogenesis is more sensitive to insulin than the inhibition of de novo glucose production (gluconeogenesis). In other words, the critical insulin level which is required to prevent ketogenesis is lower than the level required to prevent gluconeogenesis.
- The insulin deficiency is more severe in DKA compared with HHS. The residual insulin level in HHS is sufficient to minimize ketogenesis and development of ketoacidosis, but not adequate to control hyperglycemia.
The interaction between the following parameters determines the baseline insulin level in an individual patient:
- Individual’s baseline pancreatic beta cell reserve
- T1DM patients have poor pancreatic reserve
- T2DM patients have a residual pancreatic reserve
- Severity of precipitating factors (whether there’s one or multiple of the following parameters)
- States of increased insulin demand: In the presence of a catecholaminergic state, there’s more insulin demand to get back the metabolic profile to the euglycemic state. It is worth mentioning here that some stressors are strong e.g. urosepsis whereas other stressors are relatively weak such as UTI.
- Decreased pancreatic beta cell insulin secretion (i.e. pancreatic exhaustion): This happens following days-to-weeks of poor glycemic control during which pancreatic beta cells are overwhelmed with insulin hypersecretion and finally become exhausted and insulin secretion drops.
- Decreased providing insulin: patient may miss insulin dose
Click on the image to enlarge
The old maxim that patients with T1DM are prone to develop DKA whereas T2DM are prone to develop HHS is not true! In fact, this depends on the baseline insulin level. If the baseline insulin drops below the level that is required to prevent ketogenesis, then the metabolic profile shifts more toward DKA and if the insulin level is sufficient enough to prevent ketogenesis, then the metabolic profile swings more toward HHS *.
The hyperglycemic metabolic emergencies i.e. diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) are not all or none; rather it is a spectrum of metabolic derangement *. These represent extremes of the same continuum, with different levels of insulin deficiency, dehydration, ketosis, and metabolic acidosis (figure 1). Indeed, both entities may coexist in one-third of the cases.
Ketone bodies
◾️Three different ketone bodies are produced in variable amounts in ketogenic states, including:
- Acetoacetate (AcAc)
- Beta-hydroxybutyrate (BHB)
- Acetone (AC)

◾️The ratio of BHB to AcAc
- Normally the ratio of BHB to acetoacetate is 1:1.
- This ratio is altered in DKA to 3:1 and in alcoholic ketoacidosis to 10:1.
- This ratio increases further in more severe DKA and when lactic acidosis is present *.
- Of note, acetone is a neutral ketone body and does not cause acidosis or elevated anion gap. In contrast, the two other ketone bodies (AcAc and BHB) are acidic and cause ketoacidosis with a high anion gap *.
◾️Detection of ketone bodies
- Ketone bodies can be detected by:
- Nitroprusside test reagent which can be applied to urine or serum *.
- Of note, this is a qualitative method and can only detect acetoacetate and acetone (BHB is not measurable by NTP).
- The sensitivity for DKA is high (~98% *) with urinary ketones generally being > 2+ (significant ketonuria, i.e. >2+ on urine sticks is a criterion for diagnosis of DKA).
- False-negative results can also occur when urine specimens are highly acidic (for example, after the consumption of large amounts of vitamin C), or using expired or improperly stored test strips *.
- The specificity of a positive measurement of urinary ketones is low (~35% *), so a positive urinary measurement of ketones does not establish a diagnosis of DKA *.
- Direct measurement of serum BHB (capillary ketone levels), using point-of-care testing or in the hospital laboratory.
- Ketone meters are now available for rapid testing for β-hydroxybutyrate at the bedside.
- This is the gold standard for defining the presence and extent of ketoacidosis in DKA *. Blood beta-hydroxybutyrate (BOHB) concentration ≥3mmol/L is indicative of DKA *.
- Interpretation *:
- BHB is < 0.6 mmol/L: Norma.l
- 0.6-1.0 mmol/L: There is a mild ketosis condition, so adjustment of insulin therapy should be done. Among patients who initially presented with DKA, a reduction of the BHB level below <1 mM indicates resolution of the DKA *.
- 1.0-3.0 mmol/L: Moderate ketosis, medical intervention is warranted. There is a risk of progression to DKA.
- >3mmol/L: Consistent with DKA *.
- >6 mmol/L: Severe DKA.
- Nitroprusside test reagent which can be applied to urine or serum *.

◾️Correlation between ketonuria and ketonemia
- The rough correlation between the severity of ketonuria (NPT regent) and the severity of ketonemia (direct measurement of BHB) is shown in the following table.

Definition and diagnosis of DKA
According to the most recent guidelines, there are no definitive criteria for diagnosis of DKA *. Therefore it is of paramount importance to have a low threshold for considering the diagnosis in any diabetic patient who presents with hyperglycemia-related signs and symptoms (figure below).
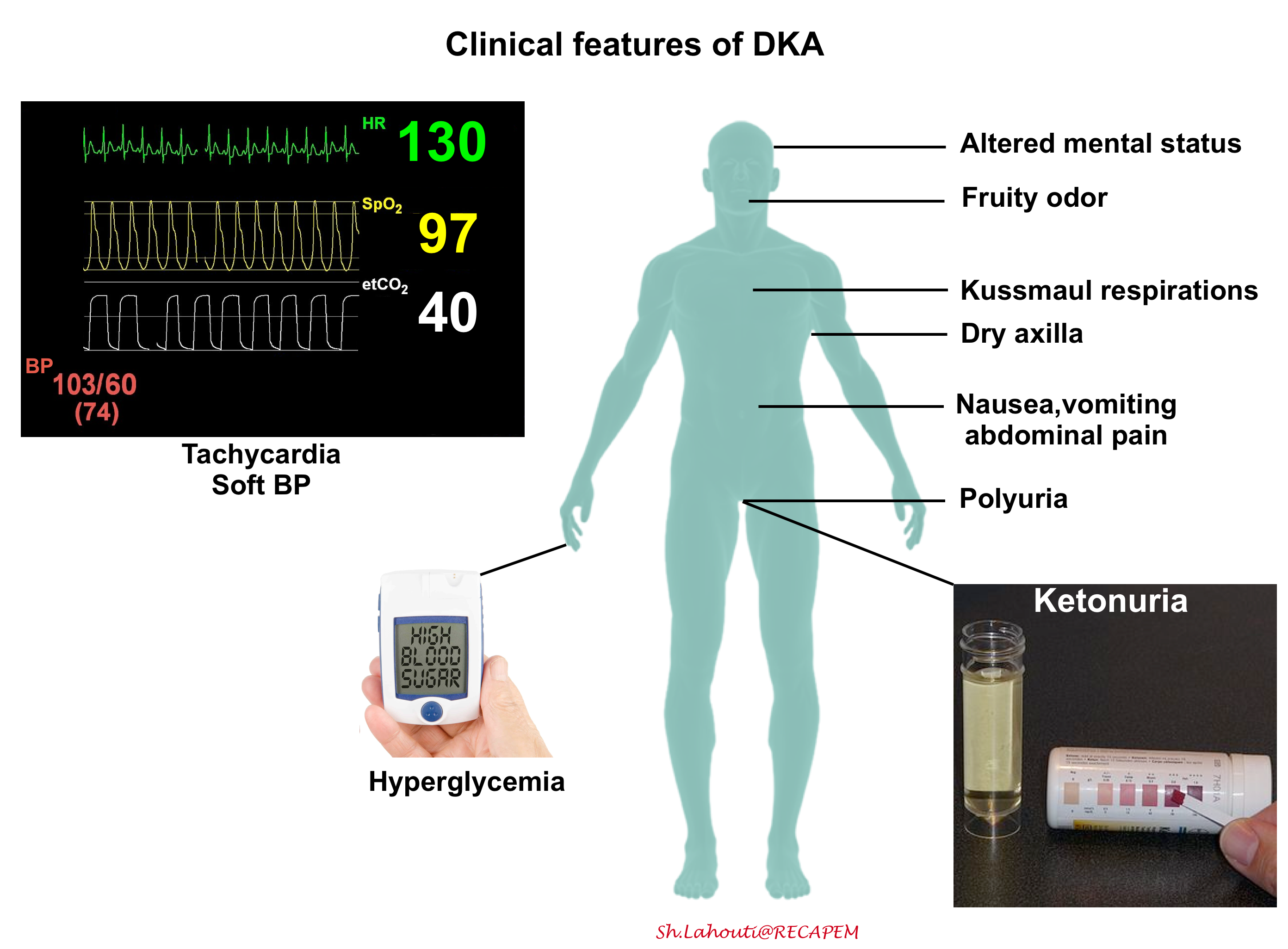
◾️Diagnosis
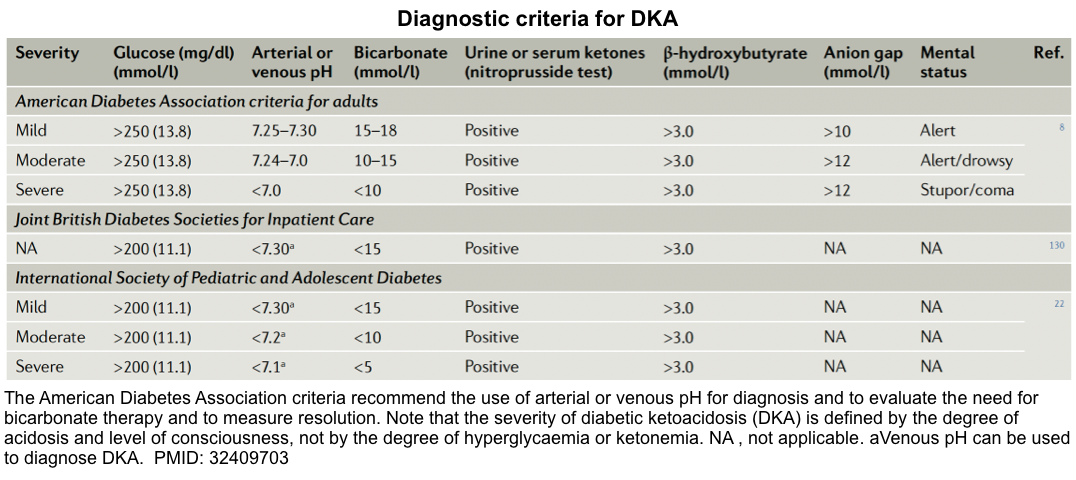
- There are no definitive criteria for the diagnosis of DKA *, and the criteria used to define DKA vary in different parts of the world (see appendix) *.
- Most patients with DKA will have the triad of hyperglycemia, high anion gap metabolic acidosis, and ketonemia or ketonuria, however, there are important exceptions:
- DKA patients can have normal glucose (euglycemic DKA, more on this here)
- DKA patients can have a normal pH and a normal bicarbonate. This usually occurs due to a combination of ketoacidosis plus metabolic alkalosis from vomiting or triggering illness *. Therefore patients with DKA can have a totally normal VBG!
▪️Elevated anion gap
- In patients with high blood glucose, DKA can be diagnosed by a high anion gap (>12) and significant elevation of BHB (>3 mM/L) *.
- The anion gap (figure 2 below) is defined as unmeasured anions and calculated as
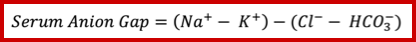
- This can be calculated simply as above and does need correction for albumin, glucose, or potassium. You do not need to correct measured sodium for hyperglycemia for the calculation of the anion gap as well.
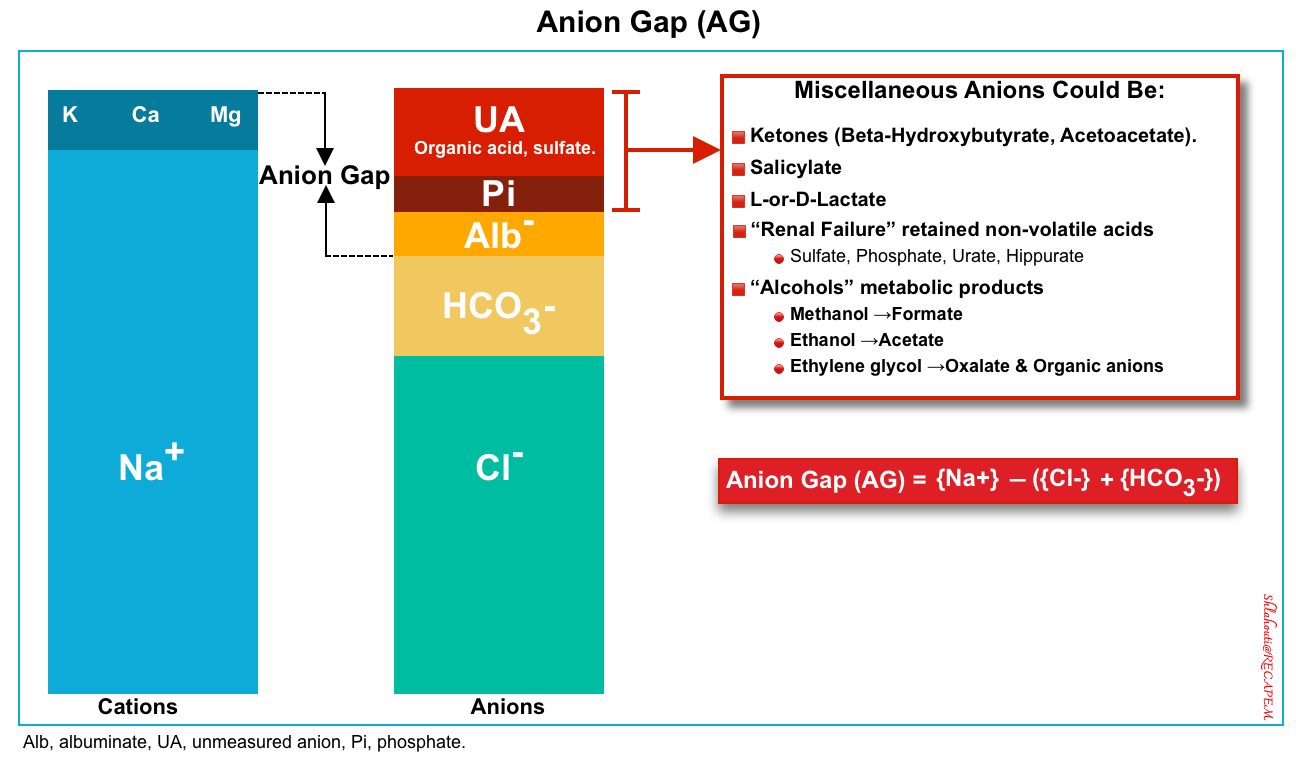
⭐️As such, DKA can be defined purely based on blood chemistry (basic metabolic profile), urine ketones, and serum BHB measurement (if necessary). VBG is not necessarily required for diagnosis of DKA (more on this here).
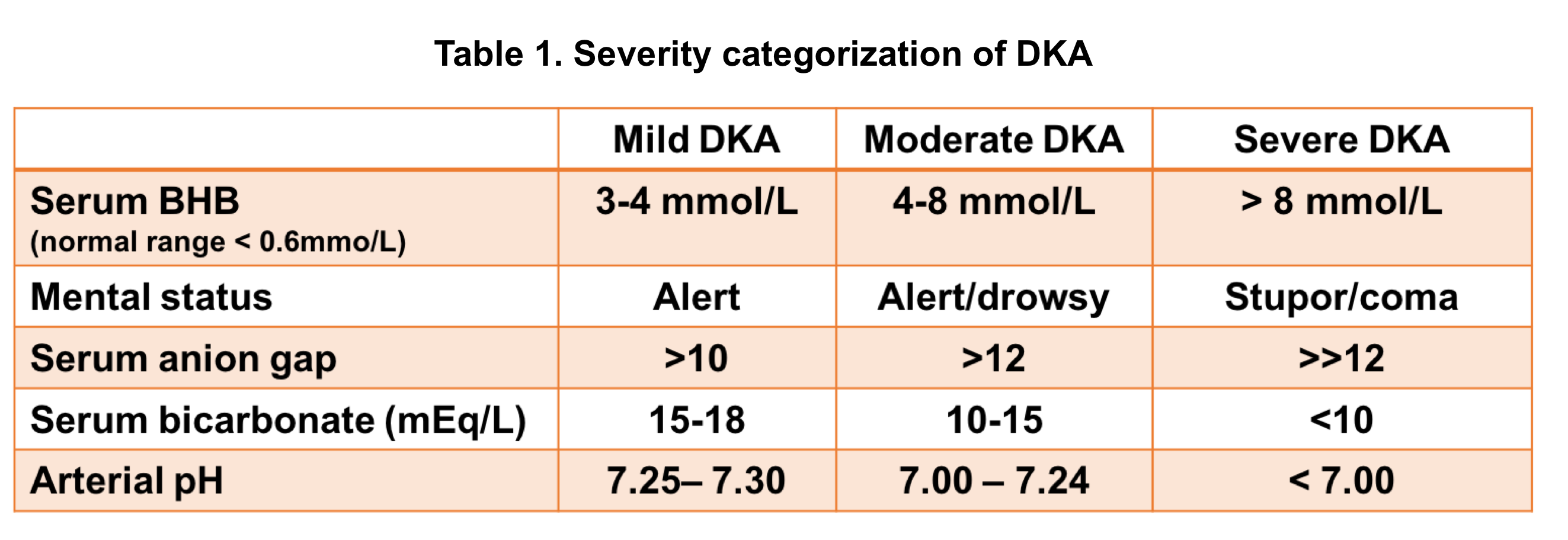
Severity of DKA
DKA severity can be classified based on certain clinical features and laboratory findings (table 1 below).
Differential diagnosis
DKA consists of the biochemical triad of hyperglycemia, ketonemia, and anion gap metabolic acidosis, each of these components can be seen in other metabolic conditions 6. The differential diagnosis of hyperglycemic crises includes other causes of ketosis, acidosis, hyperosmolality, and/or coma.
- Poor-controlled diabetes
- Stress hyperglycemia
- DKA
- HHS: In the following table, the key differences between DKA and HHS are summarized.
Hyperosmolar hyperglycemic state (HHS)
▪️The core features of pure HHS are profound hyperglycemia and altered mental status in the context of elevated serum osmolality.
◾️HHS can be defined as:
- Profound hyperglycemia (e.g. >> 600 mg/dL) should always raise concerns for HHS.
- Absence of substantial ketoacidosis (serum BHB < 3 mM or anion gap < 15mM)
- Altered mental status
- High serum osmolality > 320 mOsm (as explained below)
◾️Osmolality
- Osmolarity is a measurement of the number of particles in a solution. The serum osmolality is determined by the concentration of uncharged solute in plasma.
- In a physiologic state, the primary circulating uncharged solutes are sodium salt (mainly sodium chloride and sodium bicarbonate), glucose, and urea.
- If other solutes are present in serum at high concentrations (>5 mmol/L) such as ethanol and toxic alcohols; they will also account for the determination of serum osmolarity.
- Osmolality can be directly measured in the laboratory (based on freezing point depression). Alternatively, it can be calculated (use MDCalc).
- For diagnosis of HHS, serum osmolarity can be calculated and in this setting, it is > 320 mOsm.
◾️Consideration
- Tonicity (aka. effective serum osmolality) is a measure of the osmotic pressure gradient between two solutions across the semipermeable membrane (i.e. referring to how much the particles in a solution pull water across a semipermeable membrane). This depends on the number of effective osmoles in the solution.
- 👉Tonicity is more clinically important than osmolality because tonicity is what actually determines whether cells swell or shrink. In a physiologic state, effective serum osmolality depends on:
- Sodium: is an effective osmole, because it cannot cross the semipermeable membrane. Therefore, sodium will tend to pull water across the membrane.
- Glucose: is sometimes an effective osmole, but sometimes it’s not!
- When a patient first presents with HHS, low insulin causes glucose to stay outside the cells. This causes glucose to function as an effective osmole.
- During treatment with insulin, glucose enters the cells. At this point, glucose is an ineffective osmole.
- Ineffective osmoles: These can diffuse freely into the cells; hence do not cause pressure gradient across the cells. Examples include urea and ethanol.
⭐️The key principle to avoiding cerebral edema is to avoid rapid change in sodium or glucose concentration, especially in younger patients and those whose medical condition has been established chronically.
🔴DDx of high anion gap metabolic acidosis *
- Ketoacidosis: diabetic and alcoholic ketoacidosis
- Renal failure (uremia)
- Exogenous poisoning: Methanol, ethylene glycol, salicylate, isoniazid intoxication. More on toxic alcohol poisoning, here.
- Note that the metabolic acidosis of salicylate intoxication is related to a combination of salicylic acid, lactic acidosis, and ketoacidosis *.
- Lactic acidosis such as sepsis, cardiac arrest, liver failure, intoxication with iron, metformin, cyanide, etc.
- DKA
- Alcoholic ketoacidosis
- Blood glucose is low or normal. In the presence of low or normal glucose levels, it is less likely that it is DKA (more on this below).
- Starvation-induced ketosis
- Therapeutic ketogenic diets
- These high-fat, high-protein, low-carbohydrate diets promote ketosis by depleting carbohydrate stores, allowing the body to convert fat and protein into energy.
- The ketogenic diet does not routinely induce metabolic acidosis due to ketosis *.
- Toxin ingestion
- Toxic alcohol poisoning: Isopropanol (acetone).
- Salicylate poisoning *
- Isoniazid toxicity
- Inborn error of metabolism *
- Consider inborn errors of metabolism in children with ketosis, especially if associated with hypoglycemia (error in carbohydrate metabolism).
- Children who develop ketosis. after a longer fast may have a fatty acid oxidation disorder.
- DKA-HHS overlap (see below)
Step 1: Investigations
▪️Precipitating cause
- The goal of the initial investigation is to diagnose the metabolic hyperglycemic emergencies and the possible triggering events.
- A precipitating event can usually be identified in patients with DKA or HHS, however, DKA occasionally is the first presentation of diabetes.
- Conditions and factors associated with DKA and HHS include:
- Poor compliance with the insulin regimen, inadequate dosing, or insulin pump failure.
- Acute major illnesses such as myocardial infarction, cerebrovascular accident, bowel ischemia, sepsis, or pancreatitis.
- Infection: gastroenteritis, appendicitis, pneumonia, urinary tract infection, diabetic foot infection, etc.
- Pregnancy.
- Trauma, surgery, or any other source of physiologic stress.
- Substance abuse (e.g. cocaine*) or alcoholism.
- Medications (e.g. steroid *, atypical antipsychotics *, lithium *, sympathomimetics e.g. dobutamine, terbutaline; SGLT-2 inhibitors *, anti-calcineurin immunosuppressives *, HIV protease inhibitors *).
▪️Considerations & helpful clues
- Primary neurologic problem
- Patients with mild-moderate DKA are often alert and awake. Altered mental status can be seen in patients with severe DKA and it is commonly seen in patients with HHS (due to elevated serum osmolarity).
- 💡As a rule of thumb, serum osmolarity > 320 mOsm/kg can cause altered mental status. If the calculated serum osmolarity is < 320 mOsm/kg and the patient is altered, other etiologies for altered consciousness should be considered such as:
- Ischemic CVA, intracranial hemorrhage, cerebral vein thrombosis
- Meningitis
- Respiratory failure causing hypercapnic encephalopathy
- Cerebral edema (as a complication of treatment) when the altered mentation doesn’t improve with the treatment and so the clinical course deteriorates.
- Primary abdominal problem
- DKA can cause abdominal pain by itself, however, the provider needs to sort out whether the pain is secondary to DKA or it is due to a primary underlying abdominal problem (e.g. pancreatitis, cholangitis, appendicitis, etc.). This can be accomplished by considering the following concepts:
- Severe abdominal pain out of proportion to the severity of DKA (i.e. mild DKA) argues against DKA-induced abdominal pain *.
- Abdominal pain due to DKA should be resolved following the resolution of ketoacidosis by appropriate treatment *. If the serial abdominal examination is still revealing pain, then one must consider appropriate investigation such as abdominal CT with IV contrast.
- DKA can cause abdominal pain by itself, however, the provider needs to sort out whether the pain is secondary to DKA or it is due to a primary underlying abdominal problem (e.g. pancreatitis, cholangitis, appendicitis, etc.). This can be accomplished by considering the following concepts:
- Primary infectious trigger
- DKA itself may cause leukocytosis which is proportional to the degree of ketonemia, so an elevated WBC count is nonspecific.
- Infection is suggested by fever, bandemia (>10%), marked left-shift, or severe leukocytosis (>20.000 to 25.000) *.
- Primary hemodynamic derangement
- Patients with HHS usually have severe volume depletion with low MAP, while patients with DKA often have low-normal MAP (patients with severe DKA may present with low MAP secondary to severe acidosis). However, BP easily normalizes following initial resuscitation in patients with DKA.
- If MAP does not normalize following intravascular volume repletion, one may consider other etiologies of primary hemodynamic derangement such as sepsis, GI bleeding, and profound acidemia.
◾️Initial evaluation panel
- CBC, extended electrolytes, creatinine, BUN, as well as consideration for:
- Beta-hydroxybutyrate level & lactate, if a diagnosis of DKA is unclear.
- Cultures, urinalysis, and CXR, if an infectious trigger is suspected.
- Troponin, only if history or ECG suggests myocardial ischemia.
- If pregnancy is suspected, B-HCG is requested (pregnancy may be a trigger for DKA).
- If significant abdominal pain/tenderness:
- Lipase (note that DKA itself can increase lipase substantially) *.
- Additional investigation including abdominal CT scan may be deemed necessary in appropriate clinical settings.
- Additional workup as clinically appropriate (e.g. toxicology evaluation, CT scan to evaluate for septic focus).
Step 2: Initial resuscitation
🎯Goal: The initial goals of treatment in patients with DKA include:
- Correction of fluid deficits.
- Replacement of potassium.
- Stopping ketone production by closing the anion gap with insulin.
- Treating underlying precipitant.
⭐️The fundamental concept of treating DKA is that the issue with diabetes ketoacidosis is not hyperglycemia by itself, but rather an excess ketone body production due to a low level of circulating insulin. Therefore the cornerstone of treatment is reducing ketone production by insulin and not the correction of hyperglycemia.
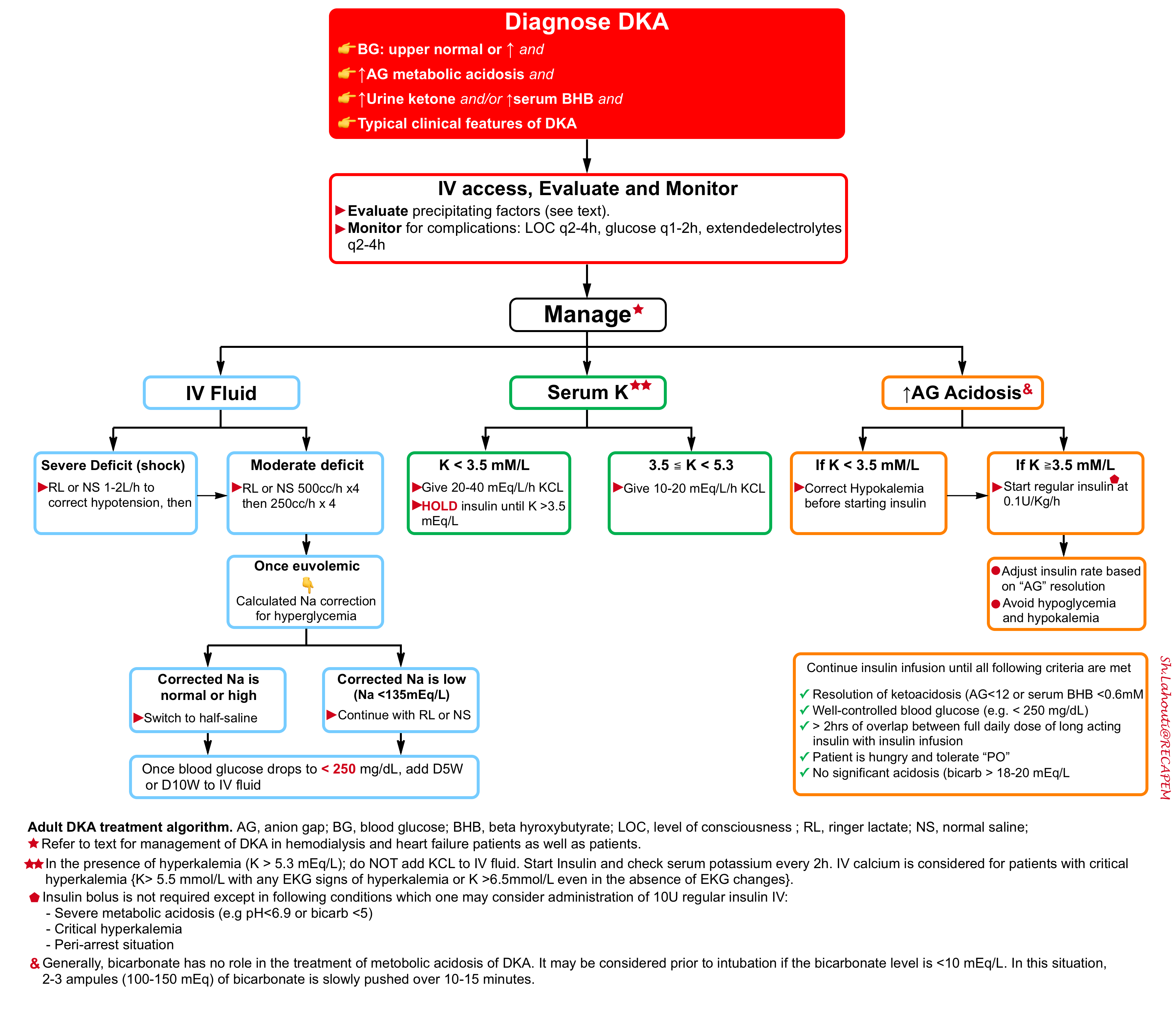
| IV Access |
- Most DKA patients can be treated with peripheral IV access, but for the sickest patients central access may be needed.
| IV Fluid |
🟥Background
- Osmotic diuresis from hyperglycemia, vomiting, and reduced oral intake make patients with DKA profoundly volume-depleted.
- Hypovolemia triggers stress hormone release which causes insulin resistance and makes hyperglycemia more difficult to treat.
- Fluid resuscitation restores intravascular volume, improves organ perfusion and renal function, and decreases insulin resistance at the tissue level.
🟥IV fluid administration
- Most patients require 2-4L of crystalloid upfront. Both NS and RL can be used.
- Balanced crystalloid is generally preferred (e.g. lactated Ringers), as this will avoid worsening the patient’s acidosis *.
- Start with 1000-1500 mL of RL over 1 hour and then adjust to the patient’s hemodynamic and electrolyte status *.
- For young DKA patients with normal cardiorenal function, if the HR is >100 bpm then they probably need more fluid.
- Ultrasound-guided fluid resuscitation is useful for patients with heart failure or patients on hemodialysis.
| Potassium |
🟥Background
- Patients with DKA and HHS have a large total body potassium deficit (~300-600 mEq/L) largely due to osmotic diuresis induced by hyperglycemia. However, the initial potassium level can be normal or even high (in 25% of patients) due to the following mechanisms:
- Intracellular fluid (ICF) shrinkage makes ICF more concentrated. Potassium will shift passively through the potassium channels.
- Solvent drag phenomenon: Refers to the passive shift of water out of the cell. This will drag potassium ions too.
- Insulin deficiency: Insulin promotes the uptake of potassium. Insulin deficiency contributes to the outward shift of potassium out of the cells.
- Note that transient hyperkalemia is not due to acidosis.
- Acidosis can indeed shift potassium outward, but this can happen for inorganic acids.
- In case of organic acidosis {ketoacidosis and lactic acidosis}, the role of acidemia is minimal. That’s why ↑K could be seen in HHS as well (in HHS there’s minimal to no acidosis).
- Keep in mind that DKA treatment will cause the potassium to drop rapidly. The potassium replacement can be managed as follows (depending on initial serum K).
🟥Potassium replacement (💡Pearls: The ticket to start insulin is the presence of serum K >3.5 mmol/L).
- If the initial serum potassium > 5.3 mmol/L: Do not add potassium to the IV fluid. Start Insulin and check serum potassium every 2h *.
- IV Calcium is considered for patients with critical hyperkalemia {K> 5.5 mmol/L with any EKG signs of hyperkalemia or K> 6.5 mmol/L even in the absence of EKG changes}. Remember that following resuscitation for DKA, the hyperkalemia will resolve promptly.
- If the initial serum potassium is 3.5 to 5.3 mmol/L: Start potassium repletion and then start insulin for treatment of ketoacidosis.
- Initial potassium 4.0 – 5.3 mmol/L: Add 10 mEq/L KCL per hour to IV fluid.
- Initial potassium 3.5 – 4.0 mmol/L: Add 20 mEq/L KCL per hour to IV fluid. Remember that you can stay on core IV Fluid (NS or RL) if no more than 20 mEq/L/h KCL is going to be given.
- If the initial serum potassium is < 3.5 mmol/L: HOLD insulin. Add KCL 20 – 40 mEq/L/h to IV fluid *,*,*. Place on continuous cardiac monitoring.
- Remember that for giving KCL at > 40 mEq/L you need to change core IV fluid to half saline.
- You can administer 40mEq/L KCL through 2 peripheral lines (20mEq/L/h via each peripheral line) or alternatively, you can give 40 mEq/L/h KCL through the central line *.
- Check serum K every 1hr.
- Insulin can be started when serum K stays > 3.5 mmol/L *.
- Continue to aggressive K repletion and shoot for a potassium >5.3 mM (targeting a high potassium level prevents you from falling behind).
| Short-acting insulin treatment |
🟥Background
- The primary problem in DKA is ketoacidosis but not hyperglycemia.
- The goal of insulting administration is to eliminate ketoacidosis and close the anion gap.
- Since glucose measurement is more feasible than serum ketones, therefore blood glucose is used as a surrogate measurement of the efficacy of insulin therapy.
- Supplemental glucose should be provided as glucose approaches normal to allow for continued insulin therapy to resolve the ketoacidosis while avoiding hypoglycemia.
- Unless the patient is hypokalemic, insulin infusion should be started immediately (see above).
🟥Short-acting insulin treatment
- IV Insulin infusion
- Preparation
- The insulin infusion is made up of 50 units of soluble human insulin in 49.5 ml 0.9% sodium chloride solution (i.e. 1 unit/ml).
- Dosing & infusion rate
- Start short-acting IV insulin at a fixed weight-based dosing of 0.1 U/kg/hr (up to a maximum of 15 units/h in morbid obesity) *.
- Higher doses of insulin (e.g. 0.2-0.3 U/kg/h) may be required in patients with severe acidosis (e.g. pH< 6.9 or bicarbonate < 5meq/L ) or those with high daily insulin requirements *.
- Should an insulin bolus dose (i.e. 10 U of IV bolus rapid-acting insulin before initiation of insulin drip) be administered before starting continuous intravenous insulin therapy?
- Insulin bolus is not required *, except in a few situations such as:
- Severe acidosis (pH < 6.9, HCO3 <5 mM).
- Critical hyperkalemia.
- Peri-arrest state.
- If setting up the insulin infusion will take > 30 minutes.
- Insulin bolus is not required *, except in a few situations such as:
- Titration
- Target blood glucose of 210-250 mg/dL *.
- The insulin infusion should be up-titrated as needed (e.g. double the initial dose) if the following “targets” are not met after the first hours of treatment:
- When serum glucose reaches 200-250 mg/dL, reduce the rapid-acting insulin infusion (but not lower than 0.05 U/Kg/h) *.
- When blood glucose < 250 mg/dL, add dextrose dextrose infusion to prevent hypoglycemia while continuing insulin infusion.
- If glucose falls < 70mg/dL, do not stop insulin infusion, but decrease by 50% (no less than 0.05U/kg/hr), provide 1 amp of D50, and switch dextrose infusion from D5 to D10.
- Patients should also be allowed to eat if it is deemed safe to do so. There is no evidence to support keeping the patient NPO.
- Preparation
- Subcutaneous insulin (for mild DKA)
- For patients with “Mild” DKA, one may consider giving rapid-acting insulin via the subcutaneous route (SC) as *:
- Initially give short-acting insulin SC at 0.3 U/Kg.
- The second dose of rapid-acting insulin SC at 0.2 U/Kg is given 1h later.
- Repeat 0.2 U/Kg rapid-acting insulin every 2 hrs.
- For patients with “Mild” DKA, one may consider giving rapid-acting insulin via the subcutaneous route (SC) as *:
| Management of severe acidosis |
🟥Most effective strategy
- The most effective strategy to manage patients with severe acidosis (e.g. pH< 6.9 or bicarbonate < 5mM) is to increase the insulin dose (usually along with the administration of additional glucose and potassium).
- Don’t wait for the insulin to arrive from the pharmacy: bolus 10 units of regular insulin IV immediately.
- Consider starting at 0.2 U/kg/hr in the sickest patients.
🟥Role of sodium bicarbonate
- Current evidence does not support replacing bicarbonate in patients with DKA*, *,*.
- There is evidence of transient worsening of ketosis and an increased need for potassium supplementation in patients who received bicarbonate*,*. Therefore routine use of bicarbonate for treatment of DKA is not recommended. The decision to give bicarbonate should be made on an individual basis depending upon hemodynamic and acid-base status.
- Experts recommend considering giving some bicarbonate before intubation if the bicarbonate level is <10 mEq/L as it may help transiently buffer the pH against the rise in CO2 that occurs during induction +/- paralysis.
| Management of refractory ketoacidosis |
🟥If the anion gap is not closing, consider the following possibilities:
- Inadequate fluid resuscitation.
- Inadequately low insulin dose.
- Malfunction of insulin infusion.
- The underlying diagnosis contributes to an anion gap that has not been addressed.
🟥Interventions if the anion gap is not closing:
- Evaluate fluid status (e.g. with ultrasonography), and provide additional crystalloid if necessary.
- Consider increasing the insulin infusion rate.
- Re-evaluate for a missed underlying diagnosis.
- Consider checking beta-hydroxybutyrate and lactate levels, to exclude an occult/worsening lactic acidosis.
Step 3: Closing the anion gap
| Monitoring |
- During the initial phase of treatment (first 6hrs), one should follow up as:
- Glucose every 1hr.
- Extended electrolyte (including Phos & Mg) every 2 hrs.
- Urine output. Glucose >250 mg/dL functions as a diuretic, so patients should produce lots of urine. Poor urine output raises concerns about shock or renal failure
| Long-acting basal insulin |
🟧Background
- Start long-acting insulin in the ED early in the treatment course *. More on this, here.
- It has been shown that early initiation of long-acting insulin facilitates transitioning off the insulin infusion, reduces the incidence of hyperglycemia, and may decrease hospital length of stay *.
- Glargine (Lantus) has a delayed onset compared to some older forms of insulin (e.g. NPH), so the traditional two-hour overlap may not work well with glargine *.
- If the patient is on a long-acting, once-daily insulin, this can be co-administered with the start of insulin infusion *.
🟧Administration
- Patients can generally be treated with their home insulin regimen (ideally a single daily dose of glargine).
- For a patient naive to insulin, a starting dose of 0.25 units/kg daily of Lantus may be given.
| Maintenance IV fluid |
- Start a maintenance fluid infusion at 250-500cc/h, once moderate-severe hypovolemia is corrected.
- The type of given fluid depends on corrected serum sodium and blood glucose.
- Keep in mind that in both DKA and HHS, the high blood glucose causes dilutional hyponatremia i.e. the measured serum sodium is falsely lower than actual values. The sodium correction for hyperglycemia can be calculated (use MDCalc) for appropriate fluid management (e.g. time to switch from NS to half-saline).
- In patients with low corrected serum sodium (e.g. <135 mEq/L), either RL or NS can be used for the replacement of volume deficit.
- Patients with a normal or high corrected sodium concentration can be switched to 0.45% sodium chloride after the first hour of fluid replacement
- Once BS drops < 250 mg/dL, dextrose should be added to the IV fluid to allow continued insulin infusion at a rate sufficient to resolve DKA while avoiding hypoglycemia. Two possible approaches are mentioned below:
- Drop and split method *
- Cut the running fluid in half e.g. from 200ml/h to 100ml/h. Add D10W infusion at an equal rate e.g. 100ml/h.The advantage of giving the components separately is that it provides you more control about adjusting the amount of sodium you are giving versus the amount of dextrose. For example, if you want to give additional D% you can up-tirate the D10W infusion (without giving the patient more sodium and causing volume overload).
- Alternatively, you may switch to D5-1/2NS at 200-250cc/h.
- Drop and split method *
| Electrolyte repletion |
🟧Potassium
- Keep potassium repletion as explained above. In patients with renal failure be more conservative with K repletion. If the patient can tolerate oral potassium replacement, it is preferred over the IV route as it is thought to have better systemic absorption.
🟧Phosphate
- Phosphate will drop during treatment, especially in patients with severe DKA. Follow the phosphate and replete if severe hypophosphatemia occurs (<1 mg/dL).
🟧Magnesium
- Maintaining a high-normal magnesium level may tend to protect against hypokalemia-induced arrhythmia if the potassium falls too low (isolated hypokalemia is usually well tolerated, whereas the combination of hypokalemia plus hypomagnesemia is more dangerous) *.
| IV bicarbonate to treat non-anion gap metabolic acidosis (NAGMA) |
🟧Background
- Following the resolution of ketoacidosis, some patients may develop NAGMA caused by 2 factors:
- Resuscitation with NS or half-normal saline
- Excretion of keto acids in urine.
- The kidney avidly excretes keto acid salts unless renal function is impaired or renal blood flow is reduced due to severe volume depletion *.
- Remember that IV fluid administration promotes normalization of intravascular volume and renal blood flow, hence promoting the excretion of keto acid salt in urine. In contrast, the institution of insulin for DKA enhances the intracellular metabolism of keto acids and regeneration of bicarbonate.
◾️Why the development of NAGMA is problematic?
- A residual acidosis will increase insulin resistance *, *, thereby increasing the risk of recurrent DKA after stopping the insulin infusion.
- If the NAGMA is severe, then it may delay the discontinuation of the insulin infusion entirely.
◾️Diagnosis

- Developing NAGMA may be revealed by the following:
- The anion gap is closing or near-closing (AG <12-18 mEq/L), but the patient’s bicarbonate remains low (e.g. < 18 mEq/L).
- The predicted final bicarbonate is a rough estimate of where the bicarbonate will end up after all the ketoacid is converted into bicarbonate (above). If the predicted final bicarbonate is falling over time to lower than 20 mM, this suggests NAGMA.
◾️Management
- Treat NAGMA with IV bicarbonate to achieve a bicarbonate level > 18-20 mEq/L before discontinuing the insulin infusion. More on this here.
- Calculate the bicarbonate deficit: MDCalc
- While the anion gap is still open, use the predicted final bicarbonate to get a rough concept of the bicarbonate deficit. Keep in mind, however, that you’re only shooting for a bicarbonate of ~20 mEq/L (not 24 mEq/L). 100-150 mEq of bicarbonate is usually adequate.
- Bicarbonate administration
- If the patient is hyponatremic, then hypertonic bicarbonate ampules can be used (each ampule contains 50 mEq sodium bicarbonate in 50 ml water).
- If the patient’s sodium is normal or elevated, then isotonic bicarbonate may be used (e.g. one liter of D5W with three ampules of bicarbonate, to generate a 150 mEq/L bicarbonate solution, infused over 3-4 hours) *.
- This will cause the glucose to increase a bit, but that can be useful in closing the anion gap (because it will trigger an increase in the insulin infusion).
Step 4: Stopping the drip
⚠️DO NOT STOP THE DRIP UNTIL ALL THE FOLLOWING CRITERIA ARE MET.
🟦Criteria for stopping insulin infusion
- Resolution of ketoacidosis (evidenced by serum anion gap < 10-12 mEq/L and or serum BHB <0.6 mM).
- An exception here is a patient with end-stage renal disease, who may chronically have an elevated anion gap due to uremia which never normalizes. In this situation, normalization of the BHB level (<0.6 mM) is a more useful way to determine that ketoacidosis has resolved.
- Glucose is reasonably well controlled (e.g. < 250 mg/dL).
- There should be at least >2 hrs of overlap of insulin drip and long-acting insulin (if Lantus has not been given yet)
- The patient should be hungry (indicating that ketoacidosis has been resolved) and tolerate “PO”.
- If the insulin infusion is stopped and the patient doesn’t eat anything or receive any IV glucose, this increases the risk of recurrent DKA.
- An exception can be made for patients with gastroenteritis or diabetic gastroparesis, who may not be hungry for a while. In this situation, the insulin infusion can be stopped, but patients should remain on low-dose intravenous glucose (e.g. D5W at 75 ml/hr). If the patient’s glucose level increases, they should be treated with PRN short-acting insulin. The ongoing administration of carbohydrates plus PRN insulin will help close the anion gap.
- The patient isn’t significantly acidotic (bicarbonate > 18-20 mEq/L) *.
- If the patient has developed NAGMA then treat with IV bicarbonate as described above.
| Start meal-associated insulin & sliding-scale insulin |
- Start meal-associated and sliding-scale insulin.
- If the patient isn’t already on a prescribed regimen of meal-associated insulin, a dose of ≈ 0.08 U/kg rapid-acting insulin per meal is reasonable. Follow glucose and titrate to effect.
- Encourage patients to eat. Carbohydrate intake (along with meal-associated and sliding-scale insulin) is important at this step to prevent recurrent DKA.
🟦Monitor for recurrence of DKA
- Check glucose level q2hrs and electrolytes ≈ 6 hrs following discontinuation of insulin drip.
- Development of progressively severe hyperglycemia may be an early sign of recurrent DKA and may pre-date the development of a widening anion gap by a few hours. If there is any concern regarding the recurrence of DKA, consider an electrolyte check to assess the widening anion gap.
| Recurrent DKA: Monitoring & Management |
🟨Causes of recurrent DKA (widening anion gap after stopping drip)
- Insulin drip was stopped despite not meeting all the above five criteria.
- Inadequate long-acting insulin dose.
- The patient isn’t eating enough (which causes insufficient meal-associated & PRN insulin doses).
- Ongoing systemic inflammation (e.g. DKA caused by infection, with persistent infection).
🟨Treatment
- Restart the insulin infusion.
- Continue long-acting insulin (consider up-titrating the dose).
- Address any reversible causes of DKA.
- Treat NAGMA to get the serum bicarbonate >20 mEq/L (this will improve insulin sensitivity).
- Severely ill patients on admission may just need a bit longer on the insulin infusion.
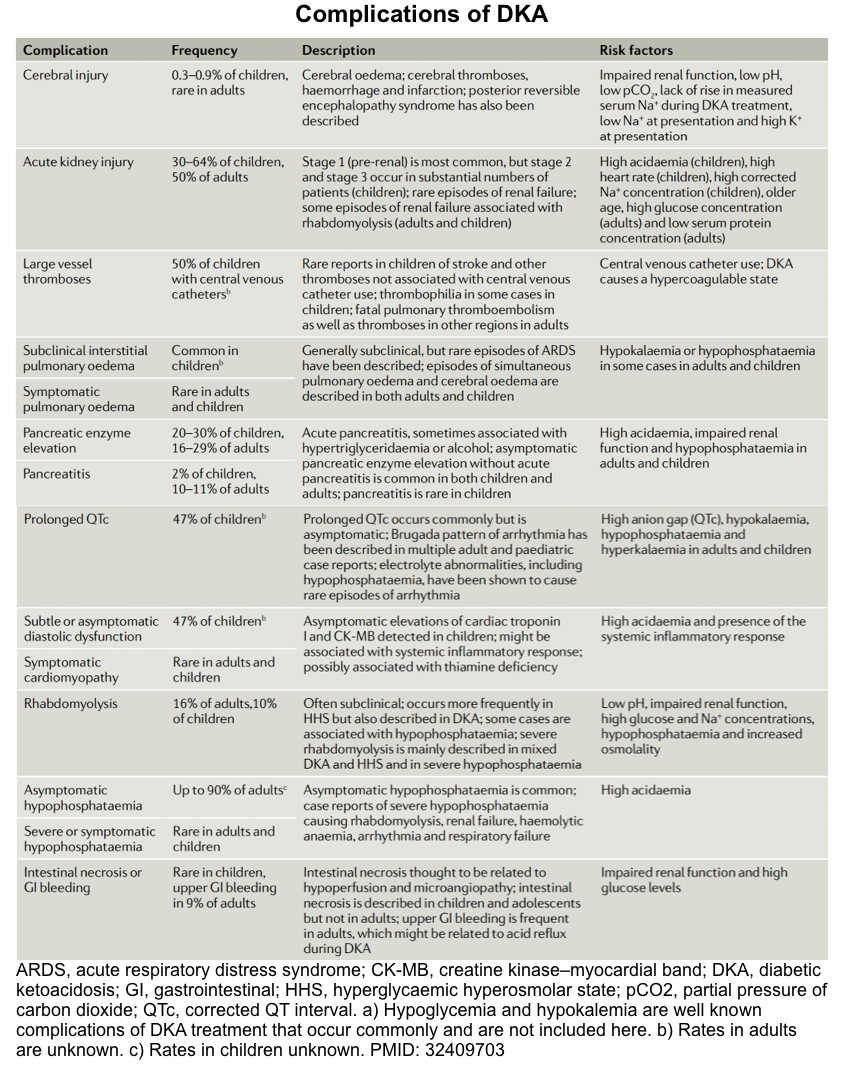
Complications
Overall view
DKA is associated with a wide range of complications. For example, hypokalemia and hypoglycemia are the most frequent complications of DKA treatment but are
generally mild and easily treated with ongoing careful biochemical monitoring (discussed above).
Other complications of DKA are summarized in the following table. In the following discussion, some important complications are discussed in detail.
Respiratory failure
◾️The effect of DKA on the respiratory system
- The presence of metabolic acidosis will normally generate a respiratory response. The reduction of serum bicarbonate and pH will result in hyperventilation and reduction in CO2, partially preventing further fall in pH and bicarbonate concentration.
- However, there is a limit to the lungs’ ability to compensate for metabolic acidosis:
- Even with serum bicarbonate concentrations below 10 meq/L, CO2 levels cannot fall < 8-12 mmHg.
- The duration of the respiratory compensation is limited by respiratory muscle fatigue.
- However, there is a limit to the lungs’ ability to compensate for metabolic acidosis:
- Respiratory pattern change with progressive metabolic acidosis.
- Initially, patients will develop tachypnea, which is ↑respiratory rate, leading to ↓CO2 concentration.
- In progressive acidosis, the respiratory pattern evolves to hyperpnea, which is ↑tidal volume.
- Finally, patients will develop a deep, fast, and agonal pattern of breathing, named Kussmaul’s respiration.
- Once patients with DKA develop Kussmaul’s respiration, they are reaching the point of respiratory muscle fatigue, and mechanical ventilation should be considered.
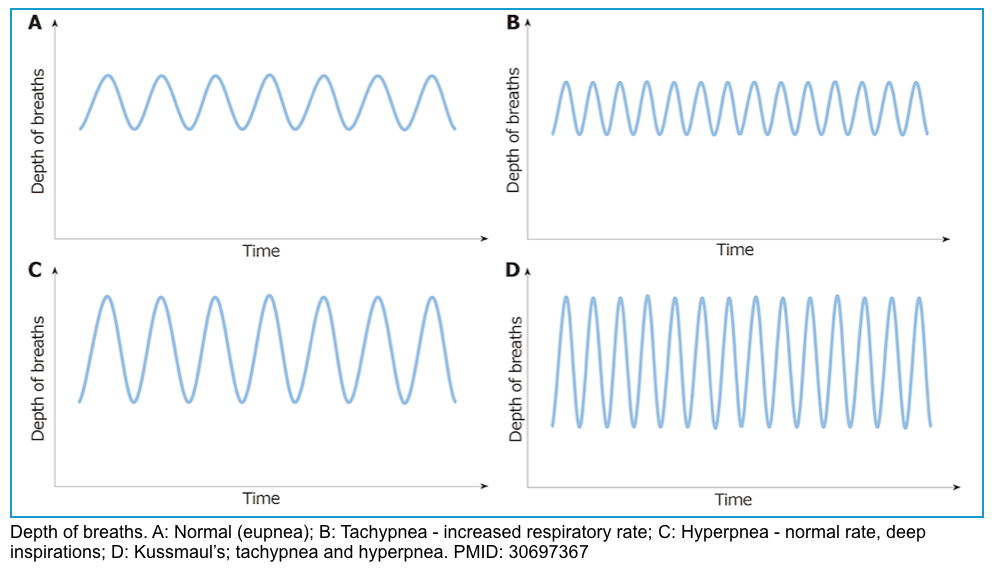
◾️Risk factors for respiratory failure
- The development of respiratory failure in DKA is associated with significantly increased morbidity and mortality *.
- Causes of respiratory failure can be detected at presentation but are usually more prevalent during treatment. These conditions can worsen respiratory function under the added stress of DKA.
- Causes of respiratory failure in DKA:
- Electrolytes abnormalities
- ↓K
- ↓Mg
- ↓PO4–
- Respiratory tract infections, e.g. pneumonia
- Clinical and radiologic features of lung infection may be masked by severe volume depletion associated with DKA, which blossoms after hydration.
- This scenario represents one condition in which the evaluation of the respiratory compensation to DKA in the ABG can offer an early sign of the presence of a condition complicating the DKA with the potential of respiratory failure *.
- Clinical and radiologic features of lung infection may be masked by severe volume depletion associated with DKA, which blossoms after hydration.
- Pulmonary edema *
- Hydrostatic (cardiogenic) due to elevated pulmonary venous pressure
- Non-hydrostatic (adult respiratory distress syndrome) due to increased pulmonary capillary permeability
- Miscellaneous conditions
- Preexisting conditions of the respiratory system
- Neuromuscular disease, e.g. Guillain- Barré syndrome.
- Central venous catheter thrombosis.
- Other chronic lung disease.
- Development of acute kidney injury during the course of DKA.
- Drugs causing respiratory depression e.g. opioids.
- Severe hypothyroidism.
- Preexisting conditions of the respiratory system
- Electrolytes abnormalities
◾️Diagnosis of respiratory failure
- Acute respiratory failure is recognized when two or more clinical features of respiratory distress are combined with one or more laboratory features listed below.
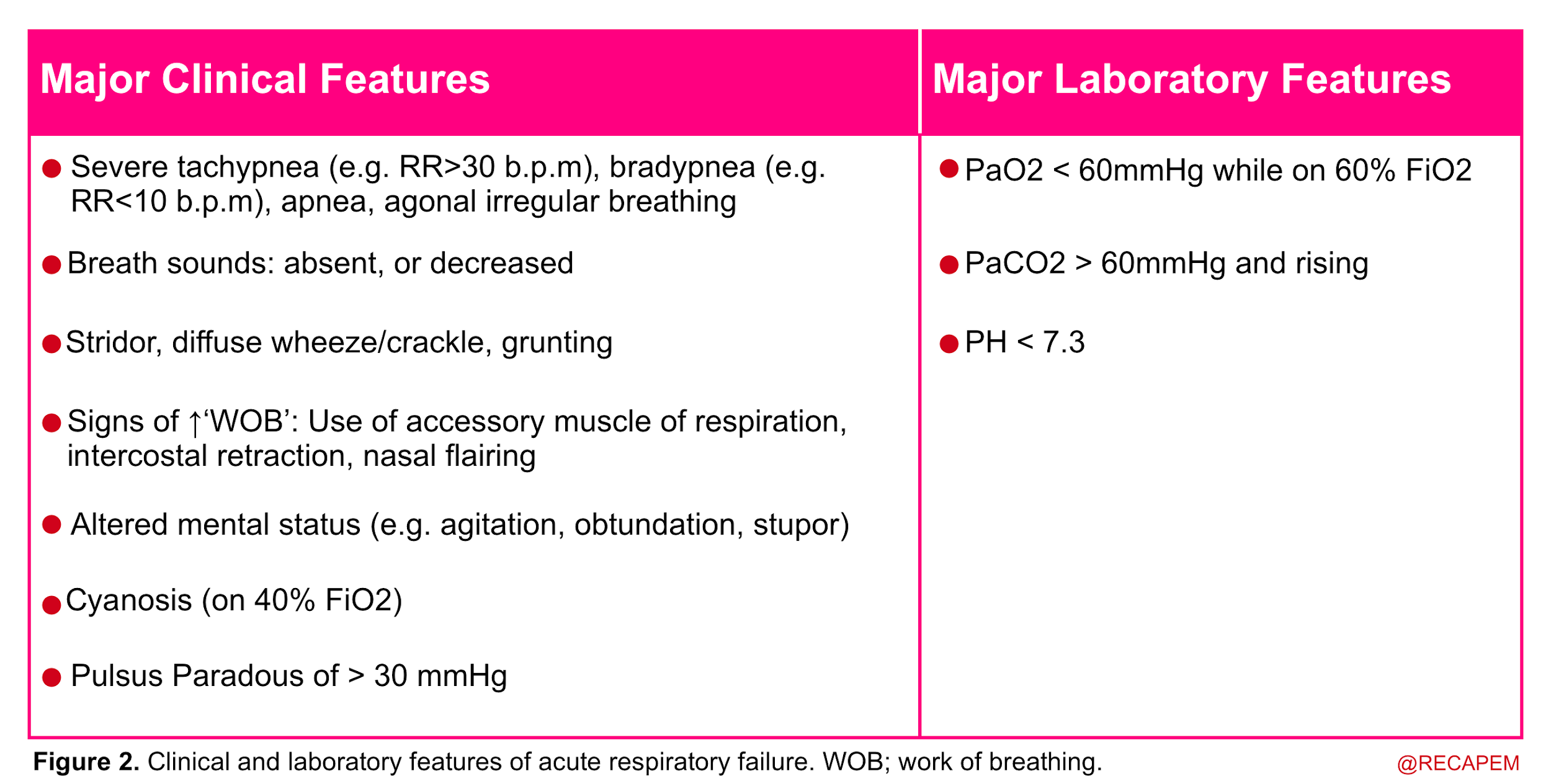
◾️Evaluation
- History and medication lists.
- Physical: Vitals? Work of breathing?
- Bedside Ultrasound/echo (more on this, here).
- Blood gas
- French guidelines recommend the measurement of arterial blood gas in every patient with decreased plasma bicarbonate level *.
- ABG to assess partial pressure of O2
- Venous blood measurements cannot detect abnormalities in the partial pressure of oxygen caused by ventilatory failure, especially in states of hypotension and tissue hypoperfusion *.
- PaCO2
- Assess the compensatory response of the respiratory system (see the following table).
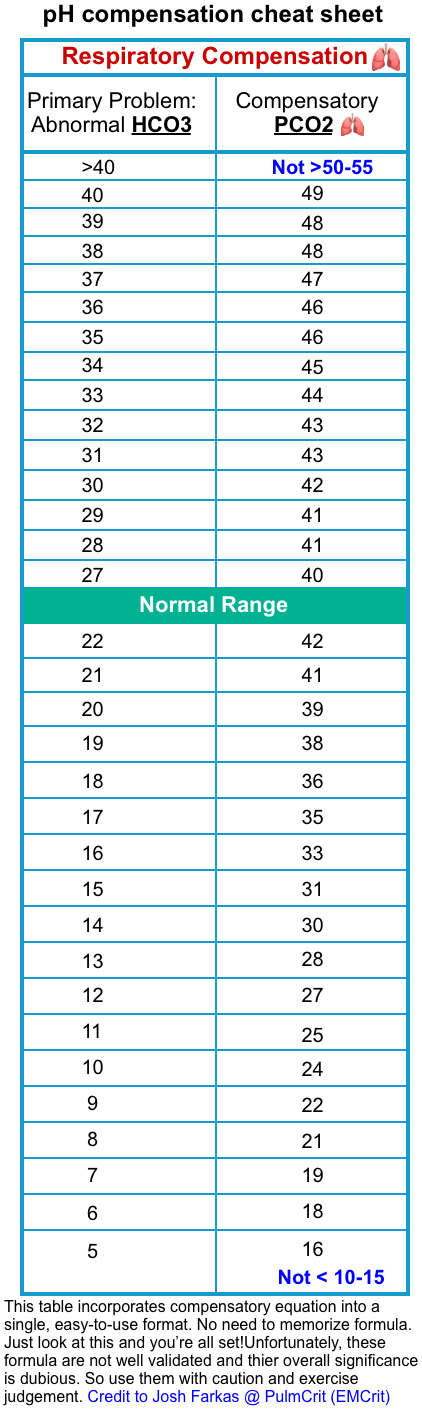
-
- If the expected PCO2 is < measured PCO2
- This could be a clue for the presence of a primary respiratory alkalosis in a patient with DKA.
- This has great importance because it often provides a clue about the presence of the following conditions *
- Sepsis.
- Respiratory distress secondary to cerebral edema.
- Other causes of respiratory alkalosis.
- If the expected PCO2 is > measured PCO2
- This could suggest respiratory failure. Such a finding should lead to a systematic search for potential causes of respiratory failure and frequent monitoring of the respiratory status of the patient including arterial blood gases *.
- If the expected PCO2 is < measured PCO2
Intubation
◾️Background
- Patients with DKA rarely have oxygenation problems, rather they may develop respiratory fatigue due to increased work of breathing.
- ⚠️Intubation of DKA patients is challenging for several reasons and possibly should be avoided *,*.
- Decompensation of acidosis and peri-intubation collapse
- Most patients have severe metabolic acidosis with compensatory respiratory alkalosis. Paralysis takes away their respiratory compensation, potentially leading to profound acidosis *.
- Hemodynamic collapse if hypovolemia and hypotension are not corrected before intubation *.
- Vomiting/aspiration: Patients with DKA often have gastroparesis and ileus which put them at high risk of vomiting and aspiration *.
- Worsening of hyperkalemia when using succinylcholine for rapid sequence intubation is likely possible.
- Increased risk of ventilator-associated lung injury
- The respiratory dynamics of hyperpnea to correct their underlying metabolic acidosis means the ventilator must equally match their large tidal volume and respiratory rate. This intrinsically puts the patient at risk for ventilator-induced lung injury and subsequent development of ARDS *.
- Decompensation of acidosis and peri-intubation collapse
- For the above reasons, unnecessary intubation should be avoided if possible.
- A high-flow nasal cannula (HFNC) is a safe way to support the patient’s breathing. By facilitating CO2 elimination, HFNC can help the patient compensate for their metabolic acidosis.
🟥Indications for intubation may include:
- A respiratory arrest or impending arrest *.
- Developing respiratory muscle fatigue (losing the ability to generate a compensatory respiratory alkalosis) *.
- Clinically obvious signs of inability to protect the airway (e.g. gurgling, inability to control secretions) *.
- Intubation before a warranted surgical procedure (e.g. patient with DKA and ongoing hemorrhage, or those with septic shock who require immediate surgery ) *.
⭐️Intubating a patient due to altered mental status is usually a mistake. The mental status should improve over several hours, therefore a careful observation is generally the best approach.
🟥If you must intubate, follow the key principles:
-
- Mitigate the risk of hemodynamic collapse: resuscitate before you intubate.
- Avoid the risk of decompensation of acidosis
- Consider for bicarbonate administration in the severe DKA (e.g. bicarb < 10 meq/l, pH < 7.0, or AG >>12). This may help transiently buffer the pH against the rise in CO2 that occurs during induction +/- paralysis *,*.
- Bicarbonate contains dissolved CO2, which the patient must blow off. In order to benefit from the bicarbonate, the patient should have enough time to blow off additional CO2 prior to intubation.
- Procedure
- Give 1 to 3 vials of 50 mL sodium bicarbonate 8.4%, IV bolus slowly push over 10 min (at least ~10 min) before intubation.
- Consider Ventilator-Assisted Pre-OXygenation (VAPOX).
- Consider for bicarbonate administration in the severe DKA (e.g. bicarb < 10 meq/l, pH < 7.0, or AG >>12). This may help transiently buffer the pH against the rise in CO2 that occurs during induction +/- paralysis *,*.
- Mitigate the risk of regurgitation and aspiration
- Perform gastric ultrasound and if distended, consider NG drainage before the intubation *.
- Consider an antiemetics.
- Meet the patient’s metabolic demand on the mechanical ventilator
- ETT Size
- Shoot for a very high minute ventilation (e.g. VM of 12-18 L/min).
- Increase tidal volume & respiratory rate to hyperventilate the patient (thus restoring respiratory compensation).
- VT (tidal volume)
- Respiratory rate
- The respiratory rate on a mechanical ventilator should be set initially similar to the patient’s compensating respiratory rate *.
- In the absence of obstructive lung disease (e.g. asthma or COPD), a respiratory rate of up to 30 breaths/min can be used safely *.
- Crank the respiratory rate as high as possible without causing autoPEEP (will often end up around ~24-28 bpm) *.
- Increase tidal volume & respiratory rate to hyperventilate the patient (thus restoring respiratory compensation).
- Mitigate the risk of hemodynamic collapse: resuscitate before you intubate.
Avoid cerebral edema
◾️Risk factors for developing cerebral edema include:
- Younger (<25 yo) patients.
- Patients with elevated baseline serum osmolarity (e.g. > 330 mOsm).
◾️Clinical presentation
- The altered mental status is associated with headache, seizure, recurrent vomiting, and/or signs of brain herniation.
- DKA-related cerebral edema occurs most commonly after several hours of DKA treatment with insulin and intravenous fluids, but can also occur at the time of presentation to the ED before treatment is administered.
◾️Best practice to avoid cerebral edema
- Avoid over-aggressive fluid administration.
- Don’t drop the glucose too fast. Avoid reducing the glucose below < 200 mg/dL.
- Replace fluid gradually. Avoid large volumes of hypotonic fluid.
- Try to use only isotonic fluids (e.g. D5 LR can be used as a source of glucose-containing IV fluid, rather than hypotonic fluids such as D10W or D5 1/2 NS).
- Avoid dropping the serum osmolality by more than 3 mMol/kg/hour or decreasing sodium by >10 mmol/24 hours
- Note that the sodium will often initially increase during resuscitation due to glucose entering the cells. This doesn’t reflect an increase in serum osmolality and doesn’t require treatment with free water. The key parameter to track is measured or calculated serum osmolarity (but not the sodium).
- Avoid unnecessary bicarbonate during treatment *.
Prevent venous thromboembolism
Patients with DKA are at relatively higher risk of VTE relative to other critically ill patients *. Provide prophylaxis against DVT unless it is contraindicated.
Special situations
DKA-HHS overlap patient
◾️As discussed earlier 25% of patients with hyperglycemia metabolic emergencies present with DKA-HHS overlap. These patients have a combination of:
- Significant ketoacidosis (serum BHB > 3 mM and substantial anion gap elevation)
- Profound hyperglycemia (e.g. blood glucose > 1000 mg/dL) with elevated serum osmolality to >> 320 mOsm.
◾️Essential concepts that should be reviewed here are:
- First: In patients with pure HHS, the disease has been established over days to weeks (this is in contrast to DKA patients whose problems have been developed more acutely). As a rule of thumb, if an abnormal state develops gradually then it may be treated gradually. This is in contrast to pure DKA patients which often require more urgent treatment to correct their metabolic problems.
- Second: remember that younger patients (< 40 yo) with DKA or HHS and those with profoundly high serum osmolality (>>320 mOsm) are at high risk for development of cerebral edema if their osmolality is rapidly decreased.
⭐️Generally, the principles of management of DKA-HHS overlap patients are the same as for DKA. The only difference is that if the patient is < 40 years old, there may be an increased risk of cerebral edema if the osmolality is decreased too rapidly. Thus, osmolality and sodium levels should be closely monitored *.
DKA in hemodialysis patient
◾️Fundamental physiologic differences
- Insulin clearance is reduced in patients with ESRD, therefore insulin requirement may be lower than in other patients.
- Osmotic diuresis secondary to hyperglycemia will not develop in ESRD patients who are anuric therefore severe volume depletion or hypokalemia is less likely to develop in these patients. In fact in some patients, hyperglycemia may osmotically pull water into the vasculature leading to hypervolemia. This can lead to volume overload with pulmonary edema, which may resolve following insulin administration (since the insulin causes a shift of glucose and water out of the vasculature and into the tissues).
- If you give excessive fluid or potassium, this will create a persistent problem that requires dialysis.
◾️Principles of Management *
- Avoid aggressive volume resuscitation
- Avoid aggressive potassium administration
- Insulin and dextrose administration
- Be conservative with insulin administration, as it may be cleared slowly. For patients who aren’t severely acidotic, it may be wise to start the insulin at a slower rate than usual (e.g. 0.05 units/kg/hour).
- For euvolemic patients, D10W may be superior to D5W to avoid causing hyponatremia. If central access happens to be available, then higher concentrations could be used (e.g. D20W).
- Oftentimes patients just need insulin but not hemodialysis
- Hemodialysis will remove keto acids and replace bicarbonate, therefore fixing the problem. However, it is not usually required since insulin alone is often adequate to improve acidosis and hyperkalemia. Hemodialysis is associated with a risk of rapid osmotic shift, especially in patients with increased serum osmolality > 330mOm/kg, it might be associated with a risk of cerebral edema.
- Even if hemodialysis fixes everything, the patient still needs insulin to prevent slipping back into DKA.
- Remember that the anion gap never normalizes in uremic patients.
- Patients on dialysis will always have an elevated anion gap, due to uremia.
- If beta-hydroxybutyrate is available, this is the best way to determine the severity of the ketoacidosis. DKA resolution may correlate with a beta-hydroxybutyrate level which is < 1 mM.
- If beta-hydroxybutyrate is not available, look at the patient’s prior anion gap values to determine a sense of the patient’s baseline (often around 12-16 mM). If the patient’s anion gap falls within this range and remains stable over time despite insulin administration, then the ketoacidosis has likely resolved.
DKA in heart failure patient
Fundamental differences
- Heart failure patients may be hypervolemic prior to developing DKA. Furthermore, they may respond less strongly to the diuretic effect of hyperglycemia. Overall, the patient may not be severely hypovolemic at baseline.
- If excessive fluid is administered, heart failure patients will tend to retain this fluid (unlike young DKA patients, who will eliminate any excess fluid via urination)
Principles of management
- Serial assessment of volume status is needed (e.g. with ultrasonography).
- Standard DKA protocols may not work. Specifically, these protocols will recommend excessive fluid administration *.
Euglycemic DKA
◾️Definition and diagnosis
- Definition: Euglycemic DKA is defined as DKA with a glucose < 250 mg/dL *.
- Diagnosis: consider the possibility of DKA whenever the anion gap is elevated or ketones are present in the urine (even if the glucose is normal) in diabetic patients taking an SGLT-2 inhibitor who present with nausea, vomiting, or shortness of breath.
- DDx: Both EDKA and alcoholic ketoacidosis have low-normal blood glucose and elevated serum anion gap. However, history provides useful clues to differentiate these two. Binge alcohol drinking suggests the diagnosis of alcoholic ketoacidosis while taking SGLT-2 inhibitors in patients with diabetes is in favor of the diagnosis of EDKA.
◾️Causes
- SGLT2 inhibitors (empagliflozin, canagliflozin, dapagliflozin) *,*
- Anything that exhausts the liver’s ability to synthesize glucose:
- Starvation, prolonged nausea/vomiting
- Pregnancy
- Hepatic failure
- Partial treatment with insulin before admission (either intentionally or unintentionally via an insulin pump)
◾️Pathogenesis
- SGLT2 inhibitor therapy in a well-compensated individual at baseline causes glucosuria, mild volume depletion, and lower serum glucose levels, associated with decreased insulin secretion *. During times of intercurrent illness and/or metabolic stress, decreased carbohydrate intake coupled with lower serum glucose levels can further depress insulin secretion. This can ultimately lead to eDKA (figure below).
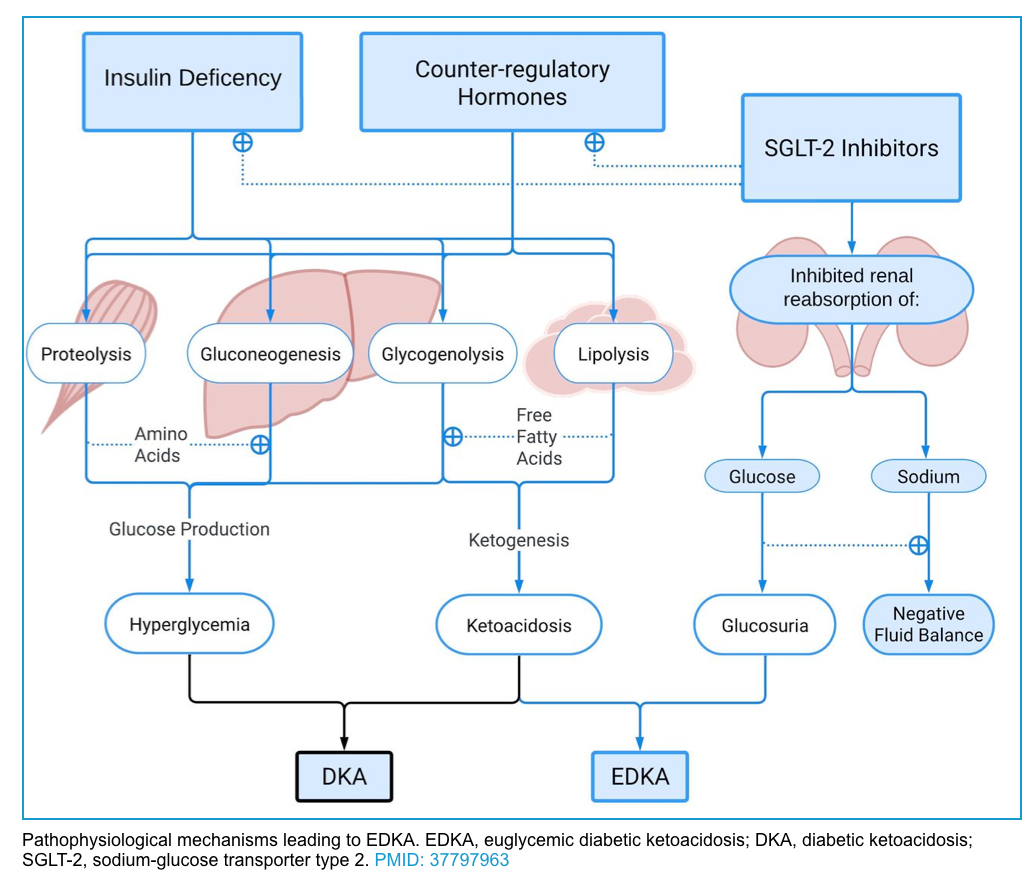
Click on the image to enlarge
◾️Treatment
- In general, treatment is similar to the usual treatment described for DKA.
- However, IV glucose will need to be started immediately (e.g. D10W or D5LR infusion). These patients will require a combination of both IV glucose plus IV insulin to resolve their ketoacidosis.
Alcoholic ketoacidosis
Alcoholic ketoacidosis usually occurs in malnourished patients with chronic alcoholism who have a history of binge alcohol ingestion. Active drinking has often stopped because of the development of abdominal pain, nausea, and vomiting. After one to two days, the patient presents to the hospital. Blood ethanol levels at this time may be low or not detectable.
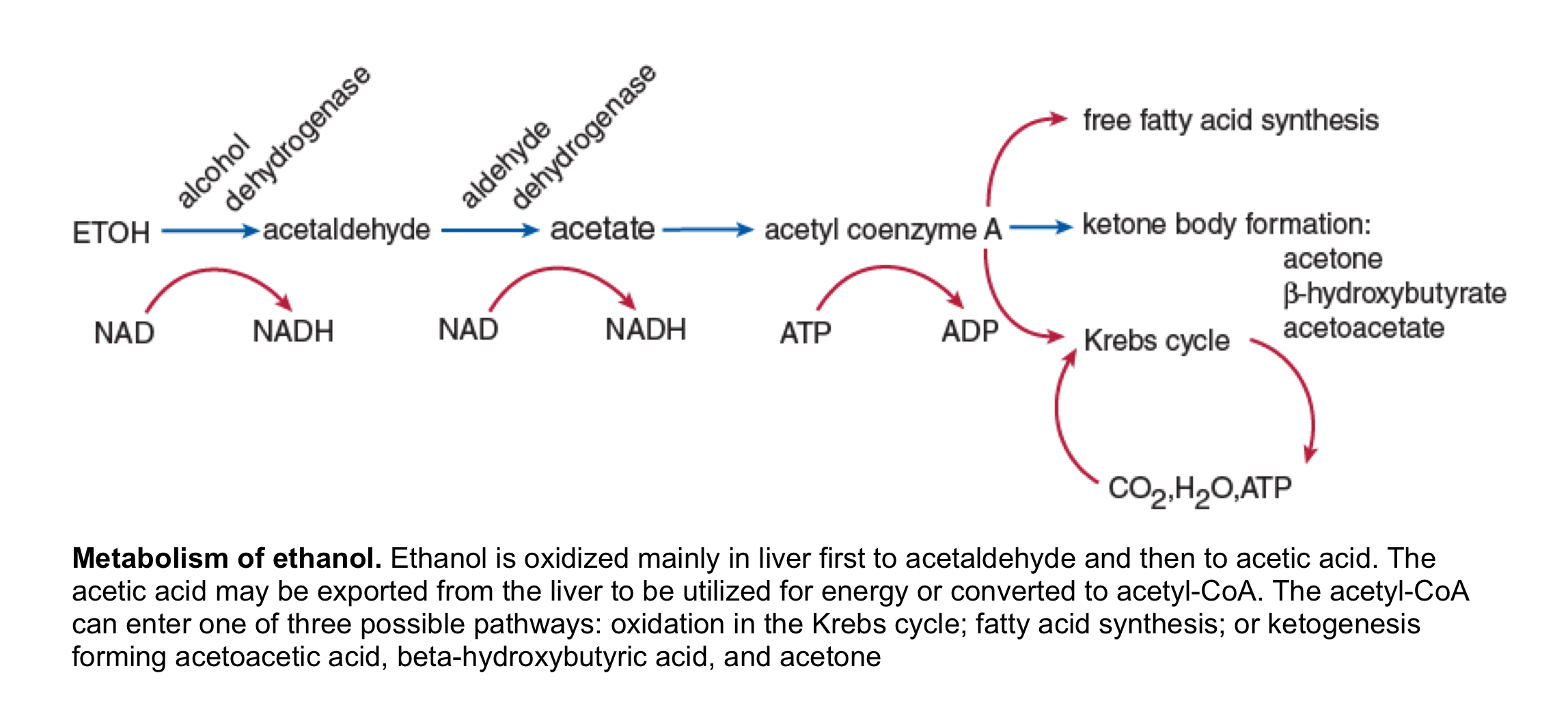
◾️Pathogenesis: the metabolism of ethanol is shown in the following figure.
Click on the image to enlarge
Following ethanol withdrawal the increased catecholamine and cortisol levels amplify the hormonal response to fasting (low insulin levels, high glucagon), causing a marked increase in lipolysis which is required for the brisk development of ketogenesis. The oxidation of ethanol to acetaldehyde and then to acetic acid each converts NAD+ to NADH. This redox shift of the NAD+/NADH ratio toward NADH has the following effects:
- It suppresses gluconeogenesis and may result in hypoglycemia.
- It increases the ratio of BHB/ acetoacetic acid. The shift from acetoacetic acid to BHB does not affect the degree of bicarbonate reduction or anion gap elevation since both acids have an identical effect on these parameters. However, it imposes a diagnostic challenge when the nitroprusside test is used.
- It favors the conversion of pyruvate to lactate. This effect is usually not important clinically, and a significant lactic acidosis in a patient with alcoholic ketoacidosis should prompt a search for other, more common disorders that are causes of a marked reduction in tissue perfusion, such as hypovolemia, heart failure, or sepsis
◾️Diagnostic criteria for alcoholic ketoacidosis
- Binge drinking results in nausea, vomiting, and decreased intake
- Low, normal, or slightly elevated serum glucose
- Wide anion gap metabolic acidosis (without alternate explanation)
- Positive serum ketones
◾️Treatment of alcoholic ketoacidosis
- Give %5 dextrose in normal saline for rehydration. Then change to %5 dextrose in half normal saline as maintenance until oral uptake resumes.
- Replace potassium and magnesium as needed.
- Optional: consider 50-100 mg thiamine and 1mg folate IV concurrent with or after glucose administration.
- Consider other causes of anion gap acidosis if the anion gap is < 20 mEq/L or fails to close with ongoing treatment.
RECAP
- Not all patients with profound hyperglycemia have DKA or HHS. Diagnosis should be made in appropriate clinical context and laboratory data.
- Avoid ruling out DKA based on a normal or near-normal VBG/bicarb or glucose. Calculate the anion gap, please!
- In clinical practice, almost 25% of patients have DKA-HHS overlap.
- The core features of DKA are high anion gap and serum BHB > 3mOm
- The main clinical features of HHS are profound hyperglycemia and elevated serum osmolality plus being unwell, especially altered mental status.
- Do not miss the underlying cause of DKA e.g. sepsis, or myocardial infarction. The most common cause of death among admitted DKA patients is not DKA, but rather its associated condition.
- DKA can cause abdominal pain by itself. However severe abdominal pain associated with mild DKA or persistence of pain despite improvement of ketoacidosis following treatment is worrisome. Make sure you perform appropriate investigations for possible underlying pathologies like pancreatitis, intra-abdominal vascular catastrophes like mesenteric vein thrombosis or intra-abdominal infection, etc.
- Hyperglycemia rarely causes mental status changes unless the serum osmolality is >320 mOsm. Thus, if the serum osmolality is <320 mOsm and mental status is significantly abnormal, look for an alternative explanation.
- If the mental status progressively worsens with initial resuscitation, consider cerebral edema, especially in a susceptible group of patients.
- Profound hypotension despite initial fluid resuscitation in DKA patients should make you consider possible underlying causes like septic shock, ongoing bleeding e.g. GI bleeding, etc.
- The main goal of DKA treatment is closing the gap, not normalizing the glucose.
- Fluid administration should precede insulin therapy to restore intravascular volume and tonicity.
- The ticket for starting insulin is serum potassium above 3.5 mEq/L. Hold insulin therapy if serum K is < 3.5.
- Start potassium replacement promptly with fluid resuscitation, if the initial potassium is < 5.3 mEq/L.
- Patients with DKA have severe potassium deficits, though serum potassium may read high superficially in some patients. This transient hyperkalemia will rapidly respond to DKA treatment.
- Do not stop the insulin infusion when glucose falls below the normal range. Patients should meet all five criteria mentioned previously before insulin infusion is stopped.
- Avoid intubation of DKA patients if possible. If you must intubate, proceed with extreme caution and preparation.
Media
Guidelines
- UK: The Management of Diabetic Ketoacidosis in Adults
- Canada: Hyperglycemic Emergencies in Adults
- International Society for Pediatric and Adolescent Diabetes (ISPAD): Clinical practice consensus guidelines on diabetic ketoacidosis and hyperglycemic hyperosmolar state
Appendix
Different diagnostic criteria for DKA
Going further
- Four DKA Pearls (PulmCrit)
- Dominating the acidosis in DKA (PulmCrit)
- Early basal insulin in DKA (PulmCrit)
- ABG/VBG unhelpful in DKA (PulmCrit)
- Hyperglycemia but not DKA (EM:RAP)
- Mild DKA (EM:RAP)
- Severe DKA (EM:RAP)
- DKA recognition and ED management (emergency medicine cases
Reference
1. Cartwright MM, Hajja W, Al-Khatib S, Hazeghazam M, Sreedhar D, Li RN, Wong-McKinstry E, Carlson RW. Toxigenic and metabolic causes of ketosis and ketoacidosis syndromes. Crit Care Clin. 2012 Oct;28(4):601-31. doi: 10.1016/j.ccc.2012.07.001. PMID: 22998993
2. Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32(7):1335-1343. doi:10.2337/dc09-9032
3. Cardoso L, Vicente N, Rodrigues D, Gomes L, Carrilho F. Controversies in the management of hyperglycaemic emergencies in adults with diabetes. Metabolism. 2017 Mar;68:43-54. doi: 10.1016/j.metabol.2016.11.010. Epub 2016 Nov 25. PMID: 28183452
4. Sheikh-Ali M, Karon BS, Basu A, Kudva YC, Muller LA, Xu J, Schwenk WF, Miles JM. Can serum beta-hydroxybutyrate be used to diagnose diabetic ketoacidosis? Diabetes Care. 2008 Apr;31(4):643-7. doi: 10.2337/dc07-1683. Epub 2008 Jan 9. PMID: 18184896.
5. Diabetes Canada Clinical Practice Guidelines Expert Committee, Goguen J, Gilbert J. Hyperglycemic Emergencies in Adults. Can J Diabetes. 2018 Apr;42 Suppl 1:S109-S114. doi: 10.1016/j.jcjd.2017.10.013. PMID: 29650082.
6. Nyenwe EA, Kitabchi AE. The evolution of diabetic ketoacidosis: An update of its etiology, pathogenesis and management. Metabolism. 2016 Apr;65(4):507-21. doi: 10.1016/j.metabol.2015.12.007. Epub 2015 Dec 19. PMID: 26975543
7. Marts LT, Hsu DJ, Clardy PF. Mind the gap. Ann Am Thorac Soc. 2014 May;11(4):671-4. doi: 10.1513/AnnalsATS.201401-033CC. PMID: 24828806
8. Nyenwe EA, Loganathan RS, Blum S, Ezuteh DO, Erani DM, Wan JY, Palace MR, Kitabchi AE. Active use of cocaine: an independent risk factor for recurrent diabetic ketoacidosis in a city hospital. Endocr Pract. 2007 Jan-Feb;13(1):22-9. doi: 10.4158/EP.13.1.22. PMID: 17360297
9. Alakkas Z, Alzaedi OA, Somannavar SS, Alfaifi A. Steroid-Induced Diabetes Ketoacidosis in an Immune Thrombocytopenia Patient: A Case Report and Literature Review. Am J Case Rep. 2020;21:e923372. Published 2020 May 18. doi:10.12659/AJCR.923372
10. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19 Suppl 1:1-93. doi: 10.2165/00023210-200519001-00001. PMID: 15998156.
11. Kondziela JR, Kaufmann MW, Klein MJ. Diabetic ketoacidosis associated with lithium: case report. J Clin Psychiatry. 1985 Nov;46(11):492-3. PMID: 3932333.
12. Taylor SI, Blau JE, Rother KI. SGLT2 Inhibitors May Predispose to Ketoacidosis. J Clin Endocrinol Metab. 2015 Aug;100(8):2849-52. doi: 10.1210/jc.2015-1884. Epub 2015 Jun 18. PMID: 26086329; PMCID: PMC4525004
13. Yoshida EM, Buczkowski AK, Sirrs SM, Elliott TG, Scudamore CH, Levin A, Tildesley HD, Landsberg DN. Post-transplant diabetic ketoacidosis–a possible consequence of immunosuppression with calcineurin inhibiting agents: a case series. Transpl Int. 2000;13(1):69-72. doi: 10.1007/s001470050011. PMID: 10743693.
14. Besson C, Jubault V, Viard JP, Pialoux G. Ketoacidosis associated with protease inhibitor therapy. AIDS. 1998 Jul 30;12(11):1399-400. doi: 10.1097/00002030-199811000-00029. PMID: 9708427
15. Umpierrez G, Freire AX. Abdominal pain in patients with hyperglycemic crises. J Crit Care. 2002 Mar;17(1):63-7. doi: 10.1053/jcrc.2002.33030. PMID: 12040551
16. Barrett EJ, Sherwin RS. Gastrointestinal manifestations of diabetic ketoacidosis. Yale J Biol Med. 1983;56(3):175-178.
17. Slovis CM, Mork VG, Slovis RJ, Bain RP. Diabetic ketoacidosis and infection: leukocyte count and differential as early predictors of serious infection. Am J Emerg Med. 1987 Jan;5(1):1-5. doi: 10.1016/0735-6757(87)90280-4. PMID: 3101715
18. Rizvi AA. Serum amylase and lipase in diabetic ketoacidosis. Diabetes Care. 2003 Nov;26(11):3193-4. doi: 10.2337/diacare.26.11.3193. PMID: 14578269
19. Tran TTT, Pease A, Wood AJ, Zajac JD, Mårtensson J, Bellomo R, Ekinci EII. Review of Evidence for Adult Diabetic Ketoacidosis Management Protocols. Front Endocrinol (Lausanne). 2017 Jun 13;8:106. doi: 10.3389/fendo.2017.00106. Erratum in: Front Endocrinol (Lausanne). 2017 Jul 31;8:185. PMID: 28659865; PMCID: PMC5468371
20. Murthy K, Harrington JT, Siegel RD. Profound hypokalemia in diabetic ketoacidosis: a therapeutic challenge. Endocr Pract. 2005 Sep-Oct;11(5):331-4. doi: 10.4158/EP.11.5.331. PMID: 16191494.
21. Andrade-Castellanos CA, Colunga-Lozano LE, Delgado-Figueroa N, Gonzalez-Padilla DA. Subcutaneous rapid-acting insulin analogues for diabetic ketoacidosis. Cochrane Database Syst Rev. 2016 Jan 21;(1):CD011281. doi: 10.1002/14651858.CD011281.pub2. PMID: 26798030
22. Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986 Dec;105(6):836-40. doi: 10.7326/0003-4819-105-6-836. PMID: 3096181.
23. Gamba G, Oseguera J, Castrejón M, Gómez-Pérez FJ. Bicarbonate therapy in severe diabetic ketoacidosis. A double blind, randomized, placebo controlled trial. Rev Invest Clin. 1991 Jul-Sep;43(3):234-8. PMID: 1667955
24. Chua HR, Schneider A, Bellomo R. Bicarbonate in diabetic ketoacidosis – a systematic review. Ann Intensive Care. 2011;1(1):23. Published 2011 Jul 6. doi:10.1186/2110-5820-1-23
25. Soler NG, Bennett MA, Dixon K, FitzGerald MG, Malins JM. Potassium balance during treatment of diabetic ketoacidosis with special reference to the use of bicarbonate. Lancet. 1972 Sep 30;2(7779):665-7. doi: 10.1016/s0140-6736(72)92083-1. PMID: 4115814.
26. Hale PJ, Crase J, Nattrass M. Metabolic effects of bicarbonate in the treatment of diabetic ketoacidosis. Br Med J (Clin Res Ed). 1984;289(6451):1035-1038. doi:10.1136/bmj.289.6451.1035
27. Fayfman M, Pasquel FJ, Umpierrez GE. Management of Hyperglycemic Crises: Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar State. Med Clin North Am. 2017 May;101(3):587-606. doi: 10.1016/j.mcna.2016.12.011. PMID: 28372715; PMCID: PMC6535398
28. Glaser N, Barnett P, McCaslin I, Nelson D, Trainor J, Louie J, Kaufman F, Quayle K, Roback M, Malley R, Kuppermann N; Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001 Jan 25;344(4):264-9. doi: 10.1056/NEJM200101253440404. PMID: 11172153.
29. Keenan CR, Murin S, White RH. High risk for venous thromboembolism in diabetics with hyperosmolar state: comparison with other acute medical illnesses. J Thromb Haemost. 2007 Jun;5(6):1185-90. doi: 10.1111/j.1538-7836.2007.02553.x. PMID: 17403099
30. Scott AR; Joint British Diabetes Societies (JBDS) for Inpatient Care; JBDS hyperosmolar hyperglycaemic guidelines group. Management of hyperosmolar hyperglycaemic state in adults with diabetes. Diabet Med. 2015 Jun;32(6):714-24. doi: 10.1111/dme.12757. PMID: 25980647.
31. Seddik AA, Bashier A, Alhadari AK, AlAlawi F, Alnour HH, Bin Hussain AA, Frankel A, Railey MJ. Challenges in management of diabetic ketoacidosis in hemodialysis patients, case presentation and review of literature. Diabetes Metab Syndr. 2019 Jul-Aug;13(4):2481-2487. doi: 10.1016/j.dsx.2019.06.022. Epub 2019 Jun 28. PMID: 31405665
32. Philip, L. and Poole, R. (2018), Double trouble: managing diabetic emergencies in patients with heart failure. Pract Diab, 35: 139-143. https://doi.org/10.1002/pdi.2181
33. Rawla P, Vellipuram AR, Bandaru SS, Pradeep Raj J. Euglycemic diabetic ketoacidosis: a diagnostic and therapeutic dilemma. Endocrinol Diabetes Metab Case Rep. 2017 Sep 4;2017:17-0081. doi: 10.1530/EDM-17-0081. PMID: 28924481; PMCID: PMC5592704
34. Wang KM, Isom RT. SGLT2 Inhibitor-Induced Euglycemic Diabetic Ketoacidosis: A Case Report. Kidney Med. 2020 Feb 22;2(2):218-221. doi: 10.1016/j.xkme.2019.12.006. PMID: 32734242; PMCID: PMC7380362.
35. Yu X, Zhang S, Zhang L. Newer Perspectives of Mechanisms for Euglycemic Diabetic Ketoacidosis. Int J Endocrinol. 2018 Oct 2;2018:7074868. doi: 10.1155/2018/7074868. PMID: 30369948; PMCID: PMC6189664.
36. PMID: 31418093. Jung B, Martinez M, Claessens YE, Darmon M, Klouche K, Lautrette A, Levraut J, Maury E, Oberlin M, Terzi N, Viglino D, Yordanov Y, Claret PG, Bigé N; Société de Réanimation de Langue Française (SRLF); Société Française de Médecine d’Urgence (SFMU). Diagnosis and management of metabolic acidosis: guidelines from a French expert panel. Ann Intensive Care. 2019 Aug 15;9(1):92. doi: 10.1186/s13613-019-0563-2.
37. Lizzo JM, Goyal A, Gupta V. Adult Diabetic Ketoacidosis. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. PMID: 32809558.
38. Vidanapathirana M. Sodium bicarbonate and intubation in severe diabetic ketoacidosis: are we too quick to dismiss them? Clin Diabetes Endocrinol. 2024 Apr 15;10(1):13. doi: 10.1186/s40842-024-00171-y. PMID: 38616273; PMCID: PMC11017618
39. Ahmed RA, Boyer TJ. Endotracheal Tube. [Updated 2023 Jul 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539747/
Add comment