

21 June, 2022. by Shahriar Lahouti.
CONTENTS
- Physiologic background
- Definition of acute liver failure
- Decompensated cirrhosis
- Acute-on-chronic liver failure
- Appendix
- Going further
- Media
- References
Physiologic background
The liver is a critical organ that is responsible for an array of functions that help support metabolism, immunity, detoxification, excretion of bile, production of clotting factors, and other proteins among other functions. Some of these functions are discussed here.
- Coagulation system proteins
- The liver manages the synthesis of nearly every plasma protein in the body, some examples include albumin, binding globulins, protein C, protein S, and all the clotting factors of the intrinsic and extrinsic pathways except for factor VIII.
- The coagulation proteins used in the extrinsic pathway (measured by PT) must be carboxylated in the liver, with a pathway that uses vitamin K, meaning an elevated PT could signal liver damage, vitamin K deficiency, or current warfarin therapy.
- While the INR is elevated in many patients with liver disease and may help to predict prognosis, a liver patient with an elevated INR is not necessarily at increased risk for bleeding, and an elevated INR in a liver patient does not rule out the possibility of thrombosis.
- The liver manages the synthesis of nearly every plasma protein in the body, some examples include albumin, binding globulins, protein C, protein S, and all the clotting factors of the intrinsic and extrinsic pathways except for factor VIII.
- Albumin. Synthesis of albumin takes place in the liver. However, hepatic albumin synthesis is not of high priority, and synthesis takes place when the body is nourished adequately. A poor nutritional state, inflammation, and exposure to hepatotoxins inhibit synthesis. In addition, albumin has a half-life of 15 to 20 days and therefore may not identify acute liver dysfunction.
- Bilirubin metabolism.
- The liver receives unconjugated bilirubin bound to albumin from the circulation. The unconjugated bilirubin then undergoes conjugation (via an enzymatic process) to conjugated bilirubin then is secreted via bile canaliculi into the bile.
- Serum-conjugated bilirubin level does not become elevated until the liver has lost 50% of its excretory capacity; while elevated bilirubin may suggest poor liver function, it cannot distinguish between hepatocellular, cholestatic, or infiltrative liver disease and may be normal or elevated in each.
- Detoxification
- The liver detoxifies ammonia to urea. In liver failure, the normal detoxification of ammonia to urea is impaired, and levels of circulating ammonia increase.
- The blood urea nitrogen concentration may not be a sensitive test to follow renal function in patients with acute liver failure since hepatic production of urea is decreased.
Liver enzymes
Basics
- These enzymes (AST, ALT, ALP, GGT) are components of hepatocytes that get released into the circulation upon hepatocyte damage.
- ALT and AST are important enzymes in gluconeogenesis, with ALT being more specific for the liver as AST is found in a variety of tissues.
- Alkaline phosphatase (ALP) can be found in the bone as well as the biliary tree, so it is not as specific, but when used in combination with the rest of the panel, it supplies evidence of hepatocellular injury. In particular, elevated ALP signals damage to the lining of the biliary tract.
- LDH. A cytoplasmic enzyme is present in all tissue in the body. Five isoenzyme forms are present in serum which can be separated by electrophoresis. The one with the lowest motility predominantly belongs to the liver. Generally, LDH is not as sensitive as aminotransferase for liver disease. It’s mainly helpful for
- Identification of hemolysis
- Distinguishing ischemic hepatitis from viral hepatitis.
Liver enzymes in liver emergencies
- The degree of elevation of liver transaminase (AST, ALT) can help guide the differential diagnosis *:
- Mild elevation: up to 5 x of normal: fatty liver, end-stage liver cirrhosis, infiltrative liver disease.
- Moderate elevation: 5-10 x of normal: alcohol-related liver disease, chronic hepatocellular and cholestatic disease.
- Severe elevation: >15 x of normal: acute liver failure including viral hepatitis, acetaminophen toxicity, shock liver, HELLP, and heat stroke.
- If massive elevation of ALT > 10.000 IU/l, then evaluate for acetaminophen toxicity or ischemic hepatopathy.
- The AST: ALT ratio of (2:1) is suggestive of alcoholic liver disease, NASH, and hepatitis C.
- Elevated ALP >x4 ULN is suggestive of cholestatic liver disease.
- In acute cholestatic liver disease, transaminase could be severely elevated. However (AST, ALT) rise of > x 25 ULN is highly suggestive of hepatocellular injury.
Definition of acute liver failure
Acute liver failure (ALF) is a life-threatening syndrome characterized by rapid deterioration of normal liver function following an acute insult in a patient with no previously known underlying chronic liver disease. Eventually, multiorgan failure may develop.
Definition. ALF is defined as a severe liver injury in a patient who meets the following requirements:
- Impaired synthetic function (INR ≥1.5) with “hepatic encephalopathy” (HE)
- Patients who present without hepatic encephalopathy are considered to have acute liver injury (but not acute liver failure).
- No underlying cirrhosis or prior liver disease.
- HE occurring within <26 weeks of the first onset of symptoms of liver dysfunction (usually jaundice) *.
👉Disease duration of >26 weeks before the onset of encephalopathy is categorized as chronic liver disease *.
👉Other signs and symptoms of hepatic dysfunction (see below) may be seen but are not required for the diagnosis of “ALF”.
Subcategorization of “ALF”
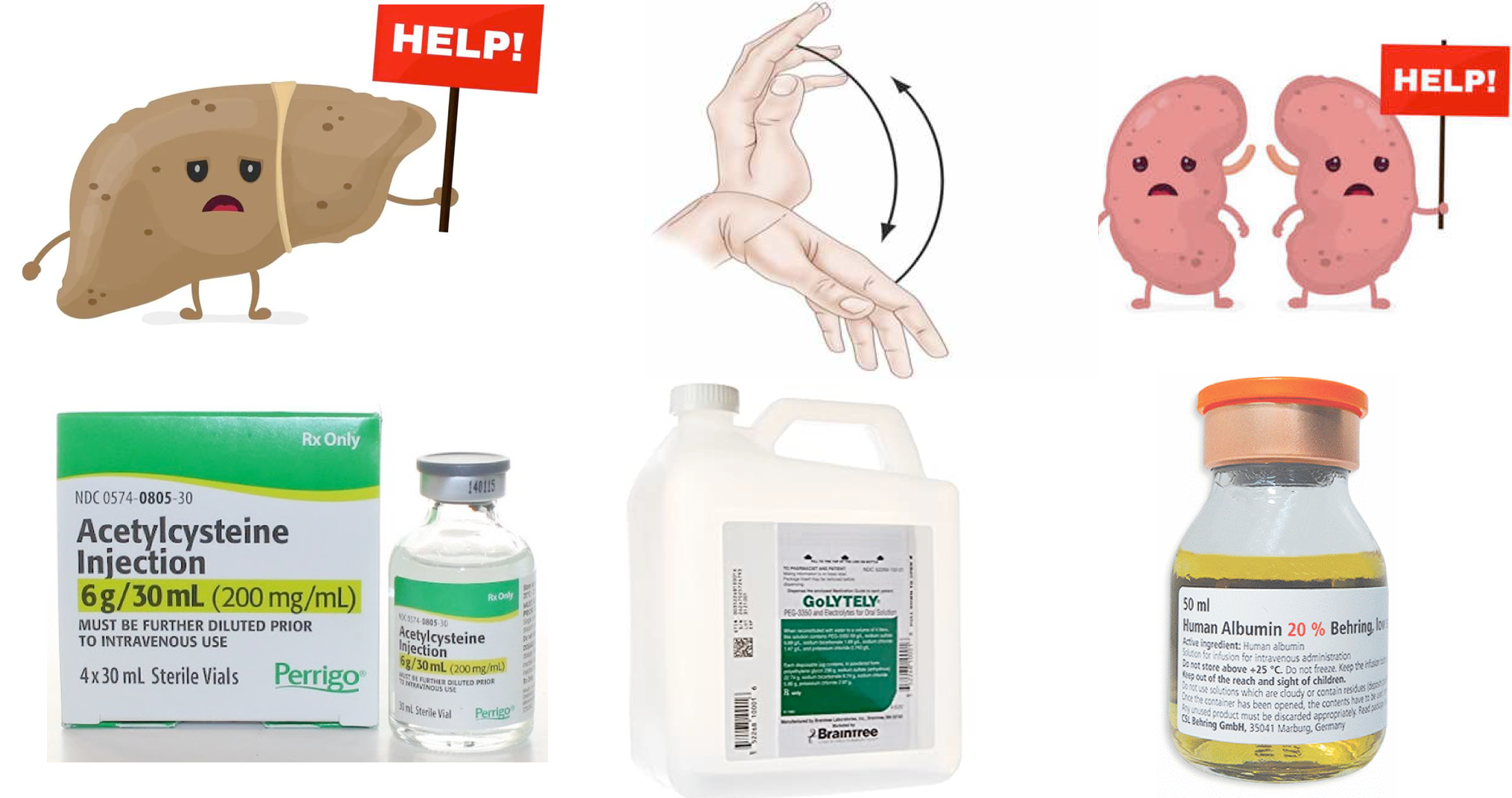
- Within the overall cohort of patients with “ALF”, subcategories exist based on the speed of progression from the initial onset of jaundice to the development of HE. Such subcategories are important because they can inform possible etiologies of ALF and help to predict outcome *. Acute liver failure can be subcategorized as
- Hyperacute: Duration between onset of jaundice to HE < 1 week.
- Overall, this has the highest risk of cerebral edema and immediate death; but also has the greatest chance of spontaneous recovery without liver transplantation (LT) if the patient survives the acute illness.
- Acute: Jaundice to HE within 1–4 weeks.
- Subacute: Duration between onset of jaundice and HE 4-26 weeks.
- This carries the lowest risk of cerebral edema and immediate death, but the likelihood of recovery without LT is low.
- Subacute cases may manifest clinically more like cirrhosis (e.g. gradual process with portal hypertension and ascites) *.
- Hyperacute: Duration between onset of jaundice to HE < 1 week.
Pathophysiology of ALF
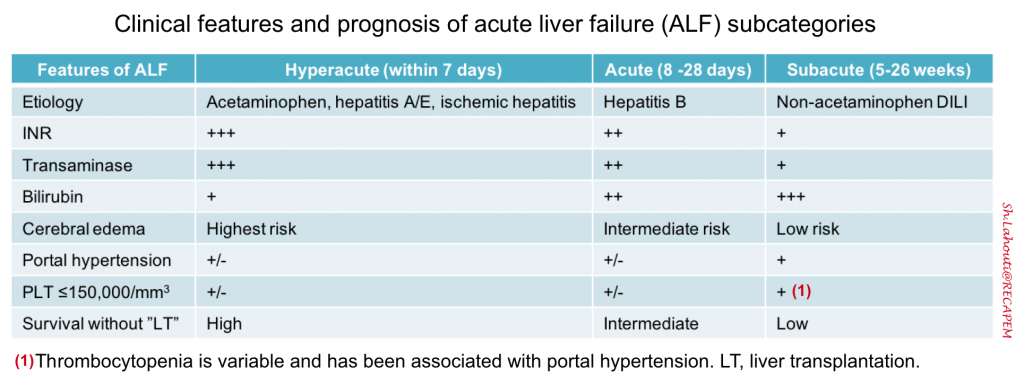
Acute liver failure is characterized by direct hepatocyte damage and necrotic or apoptotic cell damage. This results in a strong inflammatory response, both locally within the liver and systemically. Systemic inflammation contributes to the development of extrahepatic manifestations and multiple organ dysfunction syndrome (MODS). This is similar to the systemic inflammatory response syndrome, causing vasodilation and hypoperfusion *.
Hepatic encephalopathy, a hallmark feature of ALF, is characterized by altered cerebral blood flow and dysregulated cerebral autoregulation. The exact pathological basis of subsequent progression to cerebral edema and ICH is not completely understood and is likely to involve a complex interplay between systemic inflammation, circulating neurotoxins (ammonia in particular), and osmolar derangements such as hyponatremia, the end result being astrocyte swelling, brain edema, and ICH *, *.
Causes of ALF
A cause for acute liver failure can be established in approximately 60-80% of patients. Identifying the underlying cause of liver failure is important because it influences the approach to management and provides prognostic information.
-
Acetaminophen (ALF due to acetaminophen is dose-related).
-
Other drugs and toxins (idiosyncratic drug reactions). See the panel below. For a complete list of hepatotoxins, visit LivertTox.nih.gov.
- ALF due to idiosyncratic drug reactions is dose-independent. Drug-induced liver injury (DILI) usually occurs within 6 months of drug initiation.
- DILI can be classified based on clinical presentation into
- Hepatocellular (cytotoxic) injury. This causes a disproportionate elevation in the serum aminotransferases compared with the ALP *.
- Cholestatic injury. This causes a disproportionate elevation in the ALP compared with the serum aminotransferases.
- Mixed
-
Viral hepatitis
- Mostly HBV & HAV (especially reactivation of HBV in patients starting chemotherapy or immunosuppressive regimens).
- Hepatitis D can lead to “ALF” in patients with HBV infection. A patient may either acquire both viruses at the same time (coinfection) or acquire hepatitis D in the setting of preexisting chronic hepatitis B (superinfection).
- The risk of acute liver failure appears to be higher among patients who are coinfected than in those with hepatitis D superinfection or with acute hepatitis B alone.
- HEV is more common in some areas (Russia, Pakistan, Mexico, and India).
- Note that HCV alone does not appear to be a significant cause of ‘ALF’ in the absence of coinfection with HBV.
- Herpesviruses
- HSV
- CMV, EBV, and VZV in immunocompromised patients.
- Autoimmune hepatitis
- Ischemic hepatitis (aka. shock liver or hypoxic hepatitis).
- The pathogenesis of ischemic hepatitis appears to involve a ‘two-hit’ mechanism, in which a liver is at risk typically due to passive congestion, and is exposed to hemodynamic compromise *.
-
Sepsis
-
Heat stroke
-
Malignant infiltration
-
Budd–Chiari syndrome (hepatic vein thrombosis)
- Patients with prothrombotic risk factors are at high risk of developing hepatic vein thrombosis.
- Among patients with acute liver failure, the presence of hepatomegaly, right upper quadrant pain, and ascites should increase the suspicion of hepatic vein thrombosis.
-
Veno-occlusive disease
-
Wilson disease
- Pregnancy-associated.
- Acute fatty liver of pregnancy.
- Preeclampsia/HELLP syndrome.
-
Hemophagocytic lymphohistiocytosis
- Reye’s Syndrome
Drugs commonly implicated in cases of DILI.
A. Antimicrobial
1. Antibiotics: Amoxicillin, Ciprofloxacin, Doxycycline, Isoniazid, Pyrazinamide, Dapsone, Nitrofurantoin, Rifampin, Sulfonamides, Tetracycline.
2.Antiviral: Abacavir, Didanosine, Efavirenz.
3.Antifungal: Itraconazole, Ketoconazole, Terbinafine.
B. Antiepileptics: Carbamazepine, Phenytoin, Valproic acid.
C. Anti-inflammatory: Acetaminophen (paracetamol), ASA, NSAIDs.
D. Antineoplastic: Gemtuzumab
E: Herbal: Comfrey, He Shon Wu, Herbalife, Hydroxycut, Greater celandine, Kava Kava, LipoKinetix, Ma Huang.
F: Drug of abuse: Alcohol, Cocaine, MDMA (Ecstasy), Methamphetamine.
G. Antidepressant: Monoamine oxidase inhibitors, Tricyclic antidepressants.
H: Cardiovascular: Amiodarone, Methyldopa, Labetalol.
I. Poisons: Poison mushrooms (Amanita phalloides), Carbon tetrachloride.
J. Miscellaneous: Allopurinol, Disulfiram, Halothane, Isoflurane, Nicotinic acid, Propylthiouracil, Statins, Tolcapone
Clinical manifestation of liver failure
The clinical signs and symptoms of “ALF” are illustrated below. These include:
- Hepatic encephalopathy (see below)
- Jaundice
- A common misconception is that patients with ALF will be jaundiced. Jaundice may not be present despite severe decompensated acute liver failure.
- Fatigue/malaise, anorexia, nausea/vomiting, pruritus, abdominal distension, and right upper quadrant abdominal pain.
Hepatic encephalopathy
It is the essential clinical feature that differentiates ALF from acute severe hepatitis. The main feature of HE is metabolic delirium (commonly hypoactive form) with non-focal findings. Typically the severity of the HE may fluctuate throughout the course of ALF. Focal neurologic deficits, signs of ↑ ICP, and seizures may be observed with advancing grades of HE.
Grading HE *:
- Grade 1: Normal level of consciousness, changes in behavior, mild confusion, slurred speech, disordered sleep (e.g. sleep reversal), shortened attention span.
- Cerebral edema is uncommon.
- May have mild astrexis.
- Grade 2: Lethargy, moderate confusion.
- Cerebral edema is uncommon.
- Pronounced asterixis is present.
- Grade 3: Marked confusion (stupor) but arousable to painful stimuli, incoherent speech, hypo-or hyper-reflexia.
- Cerebral edema is seen in 25% of grade III HE.
- Pronounced asterixis is present.
- Grade 4: Coma, unresponsive to pain.
- Cerebral edema is common (~75%). (1)
- Asterixis is typically absent. May demonstrate decorticate or decerebrate posturing.
- Pupillary change (due to ↑ICP) may be seen as being sluggish to “fixed and dilated” (a sign typically associated with brainstem herniation) *.
(1) Cerebral edema develops in 70% to 80% of patients with grade 4 encephalopathy, irrespective of the cause of ALF. It is a unique feature of HE associated with “ALF” and a major cause of death, that is uncommon in patients with subacute ALF and chronic liver disease.
Diagnosis of HE.
- The diagnosis of HE is clinical and requires the exclusion of other causes of neurological disturbance. Virtually no test can prove the presence of HE.
- Serum ammonia is neither sensitive nor specific for diagnosis of hepatic encephalopathy *.
- Furthermore, critically ill patients may have multifactorial delirium, and HE may coexist with other causes of delirium. Therefore exploring one cause of delirium (e.g. hyponatremia) doesn’t necessarily exclude coexisting HE.
Differential diagnosis *
- Subdural hematoma
- CNS infection e.g. meningitis, encephalitis
- Nonconvulsive status epilepticus (NCSE)
- Metabolic/endocrine
- Hypoglycemia, DKA, HHS
- Electrolytes: ↑↓Na, ↑Ca
- Hyperthermia
- Wilson disease (with neurological manifestation)
- Toxicology
- Withdrawal (esp. alcohol withdrawal), opioids, benzodiazepines.
- Intoxication: sympathomimetics, salicylates.
- Organ failure
- Cardiovascular: Shock, congestive encephalopathy
- Severe infection (sepsis)
- Renal: Uremia
- Pulmonary: Hypercapnia, hypoxia
- Hematologic: TTP
- Endocrine: Thyroid storm/myxedema coma, adrenal crisis.
Investigations
-
- EEG: Only helpful when there’s clinical concern for NCSE (esp. in patients with a history of seizure).
- Optic nerve ultrasonography: Helps identify raised ICP.
- Brain CT scan: Indicated to rule out alternative causes like SDH, particularly in patients with a history of HE and/or alcoholism who are prone to falls.
- CT findings may suggest signs of cerebral edema including effacement of ventricles and cisterns, loss of sulci, and loss of differentiation between gray matter and white matter due to diffuse brain swelling *.
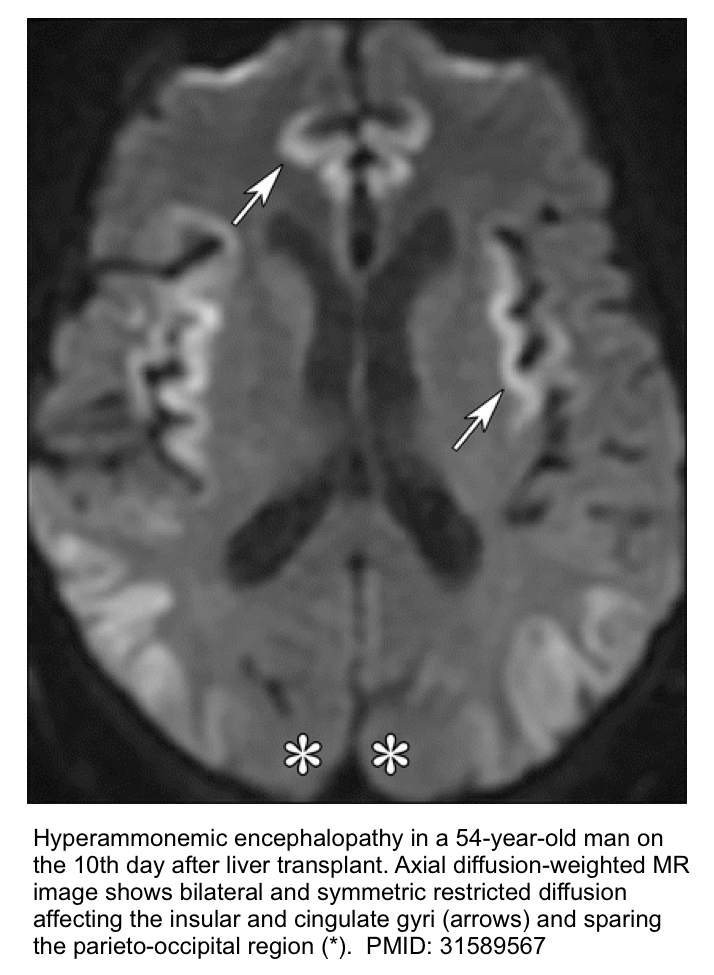
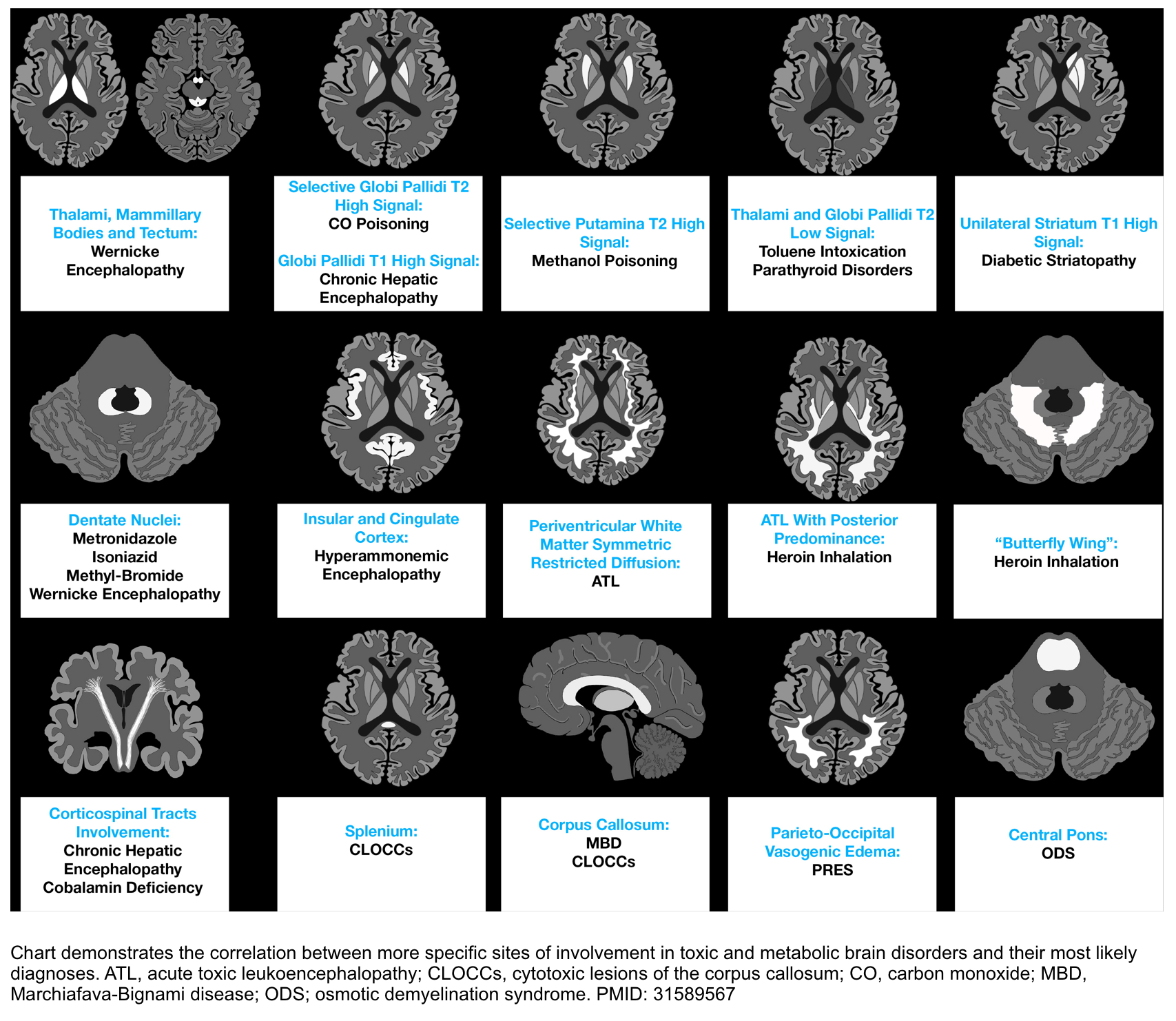
- Brain MRI.
- The classic finding is T2/FLAIR hyperintensity with diffusion restriction involving the insular gyri and cingulate gyri (with relative sparing of the occipital lobes and perirolandic region). Basal ganglia and thalami may also be involved *.
- Less severe cases may spare the cortex. These may only involve the thalami, posterior limb of the internal capsule, dorsal brainstem, and sometimes periventricular white matter*.
Lab markers of acute liver failure
- INR ≥1.5. This finding is part of the definition of acute liver failure and therefore must be present.
- Severe elevation of bilirubin level.
- Markedly elevated aminotransferase levels *
- Hyperammonemia.
- PLT count ≤150,000/mm3 is variably present and is associated with portal hypertension (esp. in subacute liver failure).
- Metabolic findings in ALF *
- Impaired renal function
- Hypoglycemia
- Lactic acidosis
Investigation
Basic investigation (to assess the severity of the disease)*
- Complete blood count and metabolic panel.
- Hepatic function panel
- Coagulation includes PT, INR, PTT, fibrinogen, and thromboelastography (TEG).
- Arterial blood gas, lactate, and ammonia levels.
Evaluation to assess complications of liver failure *
- Infection workup
- Urinalysis
- Cultures
- Chest X-ray
- Paracentesis if a substantial volume of ascites is present, to exclude spontaneous bacterial peritonitis.
- Amylase and lipase. Pancreatitis may develop in ~20% of patients with ALF (particularly in acetaminophen overdose).
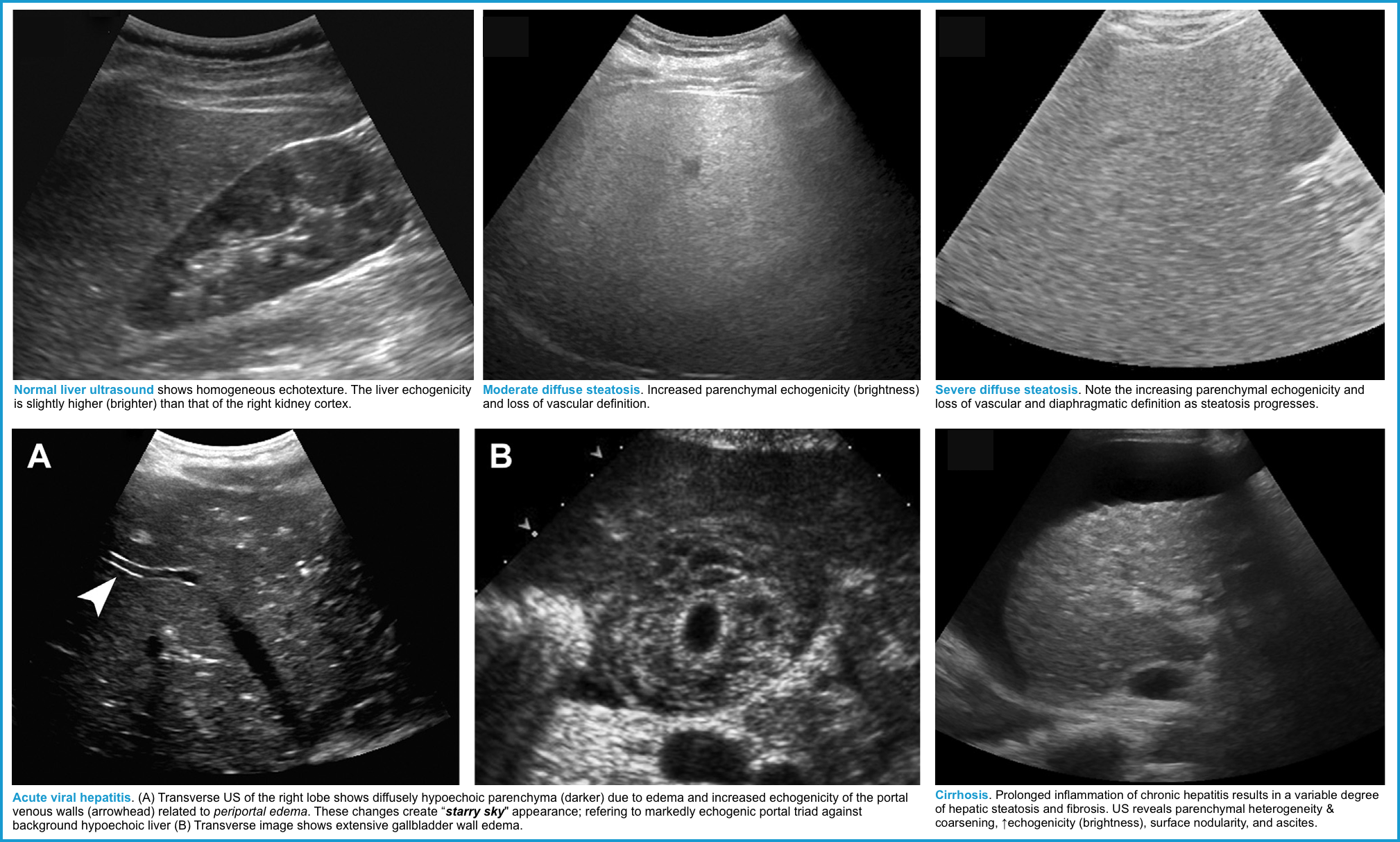
- Liver ultrasound (including Doppler study) to evaluate for Budd–Chiari syndrome, portal hypertension, hepatic steatosis, hepatic congestion, underlying cirrhosis, or malignant infiltration.
- Echocardiography: both to evaluate hemodynamics and also if the etiology is thought to be related to impaired cardiac functioning.
- Neuroimaging (CT or MRI) in patients with HE or with changes in mental status to evaluate for cerebral edema and to rule out alternative diagnoses.
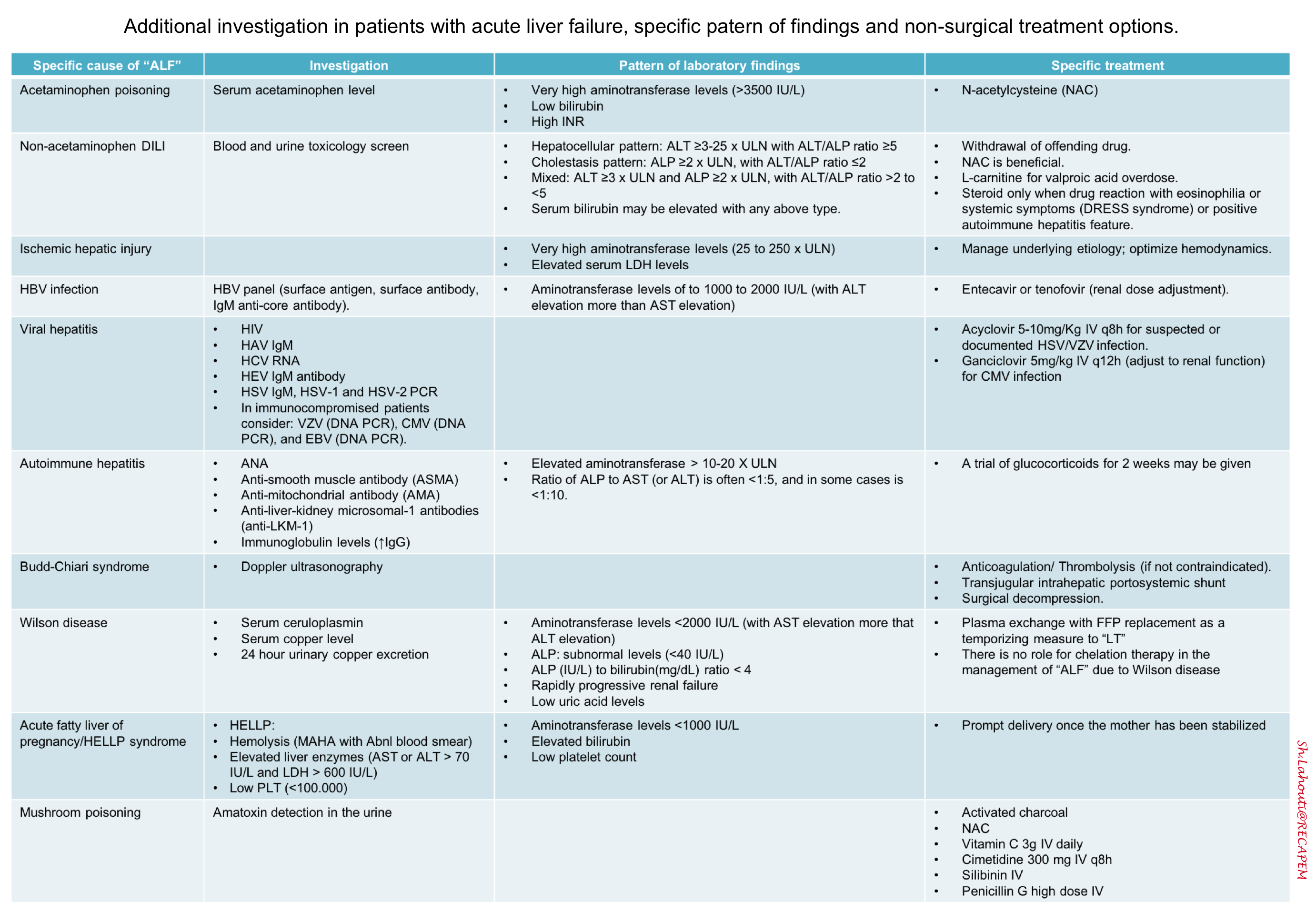
Evaluation to determine the underlying cause of liver disease (see the table below)*.
Differential diagnosis
The primary entity in the differential diagnosis of acute liver failure is severe acute hepatitis. Patients with severe acute hepatitis have jaundice and coagulopathy but lack signs of hepatic encephalopathy. Differentiating severe acute hepatitis from acute liver failure (ALF) may be challenging in alcoholic hepatitis.
- Patients with severe acute alcoholic hepatitis may present with liver failure that appears to have developed over the course of weeks to months. This is classified as acute-on-chronic liver failure (note that alcohol does not cause acute liver failure).
- Alcoholic hepatitis should be considered in patients with a history of heavy alcohol use or who have a moderate elevation of aminotransferase ( >5-10 x ULN), with an AST to ALT ratio of approximately 2:1.
- Remember that in most cases of acute liver failure, the magnitude of aminotransferase elevation is higher (e.g. >15-25 x ULN).
- Note that there is a role for corticosteroids in the treatment of alcoholic hepatitis, but not in acute liver failure.
- Alcoholic hepatitis should be considered in patients with a history of heavy alcohol use or who have a moderate elevation of aminotransferase ( >5-10 x ULN), with an AST to ALT ratio of approximately 2:1.
General management
The general management of a patient with ALF includes ensuring the patient is cared for in the proper setting, monitoring for worsening liver failure, and preventing further liver damage *.
- Admit the patient with “ALF” to the ICU.
- Consultation and timely transfer to a transplant center are crucial. This is true even for grade I and II “HE” in acute liver failure because patients can rapidly deteriorate.
Monitoring
- Serial laboratory tests are used to follow the course of a patient’s liver failure and to monitor for complications. Request for lab tests frequently as:
- Aminotransferases and bilirubin should be monitored daily.
- Coagulation panel, CBC, metabolic panels, and arterial blood gasses should be monitored more frequently (q6-8h).
- Finger sticks to monitor blood glucose levels q2-6h.
- Monitor ammonia levels because an ammonia level >150 mcg/dL has been associated with an increased risk of cerebral herniation.
- Neurological examinations at least every 2h.
- Monitor urine output hourly.
- Determine MELD score: This stratifies the severity of end-stage liver disease for transplant planning. It has poor sensitivity, but its constituents are useful in the ED to get a general sense of the severity of the disease.
Medication to be avoided in “ALF”
- Sedative medications
- In general, sedative medications (e.g. benzodiazepines, opioids, antipsychotics, antihistamines) should be avoided if possible, because patients with ALF have a severely impaired ability to clear sedatives, and the effects of sedation may mask the signs of worsening encephalopathy or cerebral edema.
- In patients who require sedative medications, use agents with extremely short half-lives in low doses (e.g. propofol, fentanyl, or dexmedetomidine) *.
- If a patient with hepatic encephalopathy can’t be rendered comfortable with low-dose propofol, it’s probably time for extubation.
- Avoid nephrotoxins. Because AKI often complicates acute liver failure, nephrotoxic drugs should be avoided.
- Analgesics
- Avoid NSAIDs. NSAIDs can cause renal impairment and even lead to hepatorenal syndrome in liver patients in addition to triggering GI bleeding.
- Opioid. Generally, it should be avoided. If opioids are required, use short-acting agents in small doses e.g. fentanyl.
- Consider IV ketamine as an alternative to opioids for analgesia in the liver patient in the ED.
- Acetaminophen should be avoided in patients with severe liver disease, ALF, and those with active alcohol use.
- In patients with mild liver disease and stable cirrhosis, a total of 2 grams of acetaminophen daily may be used daily for analgesia.
- Antiepileptic medications
- The antiepileptic medication of choice in patients with liver disease is levetiracetam.
- Carbamazepine and valproate are contraindicated in patients with severe liver disease.
- Antiemetics. Sedating antiemetics should be avoided in patients with severe liver disease. Metoclopramide and ondansetron require significant dose reduction in patients with cirrhosis.
- Antibiotics. Avoid macrolides, ciprofloxacin, nitrofurantoin, trimethoprim-sulfamethoxazole, and amoxicillin/clavulanic acid.
Identify and treat the underlying cause.
- For some causes of acute liver failure, treatment aimed at the underlying cause may prevent the need for liver transplantation and decrease mortality (see table above). Because of this, quickly determining the cause of acute liver failure is crucial.
- N-acetylcysteine (NAC). It should be given for both acetaminophen and non-acetaminophen liver failure *.
- In acetaminophen intoxication, NAC replenishes glutathione which is a crucial molecule for detoxification of acetaminophen in the liver *.
- In non–acetaminophen-related ALF (e.g. HBV), NAC seems to play a therapeutic role also as it has been shown to improve transplant-free survival *. These results may be related to other effects of NAC such as reducing the production of proinflammatory cytokines, scavenging of free radicals, changing the hepatic blood flow * as well as improving hemodynamics and oxygen use and decreasing the risk of cerebral edema *.
- The dosage is the same in either condition. See NAC Dose.
- Repeat the third dose (as a continuous infusion) until the patient is improving (and if the patient did take acetaminophen until the acetaminophen level is negative). If improvement doesn’t occur, the infusions shouldn’t be continued longer than five days.
- Defend systemic perfusion pressure.
- Patients with ALF have a hemodynamic profile similar to sepsis due to low systemic vascular resistance.
- Target a MAP > 75 mmHg *, while maintaining euvolemia.
- A higher MAP (> 75-85 mmHg) may promote cerebral perfusion in the face of elevated ICP, and may also be beneficial to renal perfusion in patients with hepatorenal physiology.
- Perform POCUS to assess volume status.
- “Only if hypovolemic“, then resuscitate with volume expanders.
- NS 0.9% is the most frequently used however a balanced crystalloid (e.g. Plasmlyte) may be considered as it has been shown in patients with acute illness that it may decrease the risk of metabolic acidosis and AKI *.
- Albumin 5% might be a preferred fluid in patients with hepatorenal physiology *. The ATTIRE trial showed that administration of albumin to target an albumin level >3 g/dL was nonbeneficial. This revealed that fluids should be titrated to “optimize hemodynamics”, rather than albumin levels *. Treat the patient but not the numbers!
- Avoid volume overload. Patients with congestive hepatopathy may be fluid-overloaded and may benefit from the judicious use of diuretics.
- “Only if hypovolemic“, then resuscitate with volume expanders.
- Vasopressor.
- ALF is a state of adrenal insufficiency. A stress dose of steroids may be considered *,*. Give hydrocortisone 50 mg IV q6h or 100 mg IV q8h.
- If suspect septic shock, obtain cultures and start empiric antibiotic therapy.
- Discontinue antihypertensive and diuretics.
- Intubate patients with worsening HE (or high-grade HE III, IV) to protect the airway and avoid hypercapnia which could worsen ICP elevation.
- Generally employ lung-protective ventilatory strategies.
- Ventilatory management, specifically related to cerebral protection, includes:
- Use of propofol as a sedative agent (this may protect from ICH and reduce the risk of seizures).
- Optimize PEEP for each individual patient. Avoid high PEEP as it may worsen ICP and hepatic congestion.
- Targeting for a PaCO2 between 35–45 mmHg.
- In the presence of intracranial hypertension short-term hyperventilation is an option, with the goal of maintaining PCO2 in the range of 25–30 mmHg. Sustained hyperventilation is prohibited to avoid cerebral vasospasm.
- In contrast to the management of acute respiratory distress syndrome, permissive hypercapnia is contraindicated (to avoid ICP elevation).
- Acute kidney injury occurs in 40–85% of patients with ALF.
- ALF associated with AKI is associated with worsening HE and poorer outcomes.
- Risk factors for developing AKI include
- Older age
- DILI due to acetaminophen, phenytoin, trimethoprim-sulfamethoxazole, or macrolides.
- The incidence of AKI is higher in ALF secondary to acetaminophen poisoning because of direct acetaminophen-mediated tubular toxicity) *.
- The incidence of AKI is higher in ALF secondary to acetaminophen poisoning because of direct acetaminophen-mediated tubular toxicity) *.
- Hypotension
- SIRS *
- Infections
- Certain etiologies: Ischemic hepatitis, acute fatty liver of pregnancy, HELLP syndrome, heat stroke, hepatitis A virus.
- Prevention and resuscitation strategy
- Avoid nephrotoxins.
- Treat Hypokalemia and hyponatremia.
- Treat AKI promptly. Consider empiric treatment of HRS with albumin plus vasopressor.
- Indication for RRT
- Classic indications e.g. severe acidosis, hyperkalemia, anuria, and/or fluid overload.
- Further indications may include the following:
- Removal of toxic substances (e.g. acetaminophen, ammonia). Early initiation of RRT in hyperammonemia (ammonia >150 uM/L) with the target to keep ammonia level <100 uM/L is recommended.
- Difficult to treat hyponatremia.
- Difficult to treat hyperthermia.
- Continuous modes of RRT are preferred over intermittent ones because the latter has been associated with an increased risk of hemodynamic instability and cerebral edema.
-
Patients with acute liver failure are at increased risk of infection sepsis and septic shock. Infection remains one of the leading causes of death in patients with ALF.
- Bacterial infection typically occurs later on during the ICU course (e.g. after >1 week *).
-
The most common sites of infection are the respiratory tract, the urinary tract, and the intravenous catheter-induced bacteremia.
- Regular periodic surveillance cultures should be performed in all patients with ALF.
-
Have a low threshold to obtain cultures, especially if:
- Presence of signs or symptoms suggesting infection (e.g. fever, cough, sputum production)
- Worsening encephalopathy especially after initial improvement
- Worsening renal function
- Elements of Systemic Inflammatory Response Syndrome (SIRS >2)
- The prophylactic antibiotic is not recommended.
-
In the presence of even subtle clinical signs and symptoms of infection or elements of SIRS; obtain cultures and start empiric antibiotic therapy.
-
Administer piperacillin-tazobactam 3.375g IV q8h) PLUS vancomycin 20-25 mg/kg IV loading dose followed by 1 g q12h.
-
Consider antifungal coverage eg, fluconazole loading dose of 200 mg IV followed by 100 mg IV q24h (esp. in high-risk patients such as those with prolonged critical care support for multiple organ failure).
-
- The coagulopathies in liver failure are complex. Most procoagulants (clotting factors) and endogenous anticoagulants are synthesized in the liver. In advanced liver diseases, levels of all these proteins are reduced to a similar extent. This leads to ‘rebalanced hemostasis’ wherein the clotting tendency is close to normal (more on this here).
- Bleeding risk is not accurately assessed by typical coagulation indices (e.g. INR). TEG is recommended to assess coagulopathy *(more on TEG here).
- Bleeding complications are rare (~<10%) in patients with ALF and reflect severe systemic inflammation *.
-
Bleeding complications were associated with lower platelet counts but not higher INRs.
-
The transfusion of any blood component was associated with an almost two-fold increase in death or need for transplantation at day 21 *.
-
- Empiric Vitamin K.
- Consider a 10 mg vitamin K IV administration (to exclude vitamin K deficiency and thereby promote accurate prognostication based on INR values) *.
- Prophylactic FFP is not recommended.
- This does not reduce the risk of significant bleeding or transfusion requirement, and can complicate trending INRs for prognosis *.
-
Coagulation factor supplementation
-
Indications:
-
Correction of coagulopathy in patients who require an invasive procedure (⚠️e.g. Lumbar puncture, liver transplant, surgical procedure, insertion of ICP monitor) *.
- Note that the following procedures are considered 👉low risk and correction of coagulopathy and thrombocytopenia is not warranted *. These include thoracentesis, paracentesis, routine upper endoscopy for variceal ligation, and central venous catheter insertion under ultrasound guidance.
-
Presence of clinically significant active bleeding
-
-
FFP, recombinant activated factor VII, or PCC
-
FFP can be given for the above indications. The main disadvantage of the FFP is that it involves high-volume administration. After a high volume is infused, there is a substantial increase in portal pressure (which can promote variceal bleeding) *.
-
Recombinant activated factor VII ( e.g. 40 μg/kg) can be considered for patients in whom FFP has failed to correct coagulopathy or those with volume overload *.
- Four-factor prothrombin complex concentrates (PCC). The main advantage of PCC is that it involves less volume administration, which may be especially useful in patients with variceal hemorrhage (wherein volume loading may increase the pressure within the varix, thereby promoting further bleeding). PCC may be more effective at restoring thrombin generation, compared to fresh frozen plasma*.
-
- Fibrinogen supplementation is reasonable (for the above indications) when the fibrinogen level is <~150 mg/dL. Either cryoprecipitate or fibrinogen concentrates may be used *.
-
- PLT transfusion. Maintain a platelet threshold of
- >10,000/mm3 in non-bleeding patients.
- >50,000/mm3 in patients with significant bleeding *.
- >50,000-70,000/mm3 in patients undergoing invasive procedures.
- Control infection and optimize renal function *
- Active infection may cause the release of endothelial-derived “heparinoids,” which can have an anticoagulant effect.
- Superimposed renal failure is associated with uremic platelet dysfunction, volume issues, and changes in the hemostatic cascade.
- DVT prophylaxis
- Despite the elevated INR, patients in ALF have a tendency towards coagulation (even more than patients with cirrhosis).
- In the absence of bleeding, routine DVT prophylaxis in hospitalized patients with cirrhosis is generally recommended *.
- In patients with cirrhosis and the following conditions, DVT prophylaxis may be contraindicated: active bleeding, severe thrombocytopenia (~<20.000 u/L), fibrinogen <80 mg/dL, TEG showing a pathologically prolonged R-time.
- When in doubt (e.g. patients with profoundly elevated INR), TEG may be helpful to understand the patient’s coagulation balance.
- Stress ulcer prophylaxis should be given for all patients.
- Avoid constipation, with a low threshold to initiate lactulose as the cathartic agent of choice.
- Nutritional support:
- Nutritional support is a vital component in the treatment of acute liver failure and should be initiated early. It is required to prevent catabolism of body stores of proteins and it may decrease the risk of gastrointestinal bleeding from stress ulceration in critically ill patients.
- To prevent protein catabolism, severe protein restrictions should be avoided *. Standard protein targets can generally be utilized (1.2-2 grams/kg IBW/day).
- Monitoring ammonia may be useful to avoid exacerbation of hyperammonemia.
- For marked hyperammonemia (e.g. >150 uM/L) and high-grade encephalopathy, temporarily suspend feeding, until ammonia levels are controlled.
-
Treat hypoglycemia with a glucose infusion.
-
If the level is <100 mg/dL, begin a 10% dextrose infusion via the peripheral line or higher concentration via the central line; with a goal glucose level of >100 and <180 mg/dL *.
-
- Correct electrolyte disturbances.
- Avoid hyponatremia.
- Target a high-normal sodium level (140-145 mmol).
- Avoid large volume infusion of hypotonic fluids.
- Infusion of hypertonic saline to maintain serum sodium between 145 and 155 mmol has been shown to be associated with reduced ICP.
- RRT can be utilized to correct hyponatremia.
-
If the patient is acidotic, consider a sodium bicarbonate infusion.
👉Hepatic encephalopathy
- Hepatic encephalopathy is one of the defining characteristics of acute liver failure and is frequently associated with cerebral edema. In this context, it is far more dangerous than hepatic encephalopathy seen in chronic cirrhosis which isn’t associated with cerebral edema or herniation.
- Principles of management of hepatic encephalopathy
- If possible, avoid sedating medications (esp. those with prolonged half-life) that may confound the picture or exacerbate encephalopathy.
- Correct electrolyte abnormalities esp. hypokalemia, and hyponatremia.
- Although lactulose and rifaximin are mainstays of managing HE in chronic liver disease (see below), there is little demonstrable benefit in ALF *.
- Perform intubation for high-grade HE (III, IV) to prevent aspiration.
- Correct hyperammonemia. Early RRT when ammonia levels are >150 uM/L may improve survival.
👉Cerebral edema
- The cerebral perfusion pressure (CPP) is the difference between mean arterial pressure and ICP.
- The consequences of cerebral edema include ICP elevation, brain ischemia and hypoxia, and brainstem herniation, which are the most common causes of death in “ALF”.
- Signs and symptoms
- Severe encephalopathy is the main sign of intracranial hypertension.
- Other findings may include hypertension, bradycardia, abnormal respiratory patterns (Cushing’s Triad), mydriasis, and decerebrate posturing.
- Risk factors for ICP elevation *
- Shorter duration from jaundice to encephalopathy (hyperacute ALF).
- Younger ages
- Renal dysfunction (more severe renal injury results in greater ammonia retention)
- Hypotension requiring vasopressor support.
- Ammonia levels*
- >100 uM/L may predict the onset of severe hepatic encephalopathy with 70% accuracy.
- >150-200 uM/L correlates with risk of herniation.
- Diagnostic studies
- Optic nerve diameter measured by ultrasonography or brain CT is useful.
- Brain CT is not sensitive. However, eventually, diffuse narrowing of sulci and compression and effacement of ventricles and cisterns becomes apparent.
- Transcranial Doppler ultrasound. Is another method being studied for the detection of ICP elevation, however, it requires expertise.
- Invasive ICP monitoring. It may be used for those at greatest risk for cerebral edema, namely patients with grade IV encephalopathy or in patients with grade III encephalopathy that is rapidly progressing.
- Preventive measures for ICP elevation *
- Minimize venous congestion. Elevate the head of the patient’s bed to 30-45 degrees.
- However, bed elevation can also reduce cerebral perfusion. Thus, experts have recommended not raising the head of the bed if the cerebral perfusion pressure cannot be maintained at an appropriate level (i.e. 50 mmHg) with bed elevation
- Minimize patient agitation/stimulation.
- Patients should be placed in an environment with minimal sensory stimulation.
- Avoid or minimize procedures that make the patient agitated.
- Nasogastric tube placement can cause gagging and thus should generally be performed only in patients who are intubated and sedated.
- Similarly, endotracheal suction should be minimized.
- Maintain euglycemia and normothermia.
- Target a serum Na level at 140-145 mmol/L.
- Maintain PCO2 within a low-normal level.
- Minimize venous congestion. Elevate the head of the patient’s bed to 30-45 degrees.
- Treatment of ICP elevation
- Goal: To reduce the ICP <20-25 mmHg and to maintain cerebral perfusion pressure (CPP) >50-60 mmHg.
- Consider bolus hypertonic saline (250 ml of 3% NaCl) or 20% mannitol (1g/kg bolus) IV given over 20 min. Doses can be repeated once or twice as long as serum osmolality remains < 320 mOsmol/L.
- Ensure optimal sedation with propofol (it may improve ICP and also prevent seizures).
- Consider a short period of hyperventilation, to reduce PaCO2 to 25–30 mmHg.
- Consider early RRT (in addition to classic indications) for the following conditions:
- Hyperammonemia: RRT when ammonia levels are >150 uM/L may be associated with improved survival *
- Hyponatremia
- Increased serum osmolarity.
- Treatment of seizure
- Seizures in patients with ALF should be treated promptly because seizure activity increases ICP and may cause cerebral hypoxia.
- Subclinical seizure is often present in grade III-IV encephalopathy but may be difficult to detect if patients are intubated and receiving paralytics. Therefore have a low threshold for obtaining video EEG.
- Prophylactic antiepileptic agents are generally not recommended.
- Among intubated patients (who require sedation), propofol is preferred with the advantage of providing both sedative and antiepileptic activity.
- Phenytoin has traditionally been used as the medication of choice; however, agents without risk of hepatotoxicity and more easily achieved therapeutic levels such as levetiracetam are now more frequently utilized.
Decompensated cirrhosis
Definition of cirrhosis
- Chronic liver disease (CLD) is a progressive deterioration of liver functions for more than 6 months.
- Cirrhosis is a final stage of chronic liver disease with different etiologies. The clinical manifestations of cirrhosis are related to portal hypertension and hepatic dysfunction, progressing to liver failure as a result of progressive fibrosis and destruction of lobular and vascular architecture.
Complications of cirrhosis
Patients with cirrhosis are susceptible to a variety of complications. By and large, these complications are produced by two sets of ongoing pathophysiologic processes:
- Progressive loss of liver functional reserve
- This can lead to hyperbilirubinemia (jaundice), coagulopathy, hyperammonemia (encephalopathy), and hypoalbuminemia.
- Portal hypertension.
- Distortion of liver microcirculation by fibrosis, nodules, and vascular occlusion causes resistance to portal blood flow, which leads to the development of portal hypertension.
- Increased pressure within the portal venous system can lead to congestive splenomegaly (causing thrombocytopenia) and the formation of venous collaterals (varices) as well as circulatory, vascular, functional, and biochemical abnormalities. These contribute to the pathogenesis of portal hypertension complications including:
- Variceal hemorrhage
- Portal hypertensive gastropathy
- Ascites
- Spontaneous bacterial peritonitis (SBP)
- Hepatorenal syndrome (HRS)
- Hepatopulmonary syndrome
- Hepatic hydrothorax
- Portopulmonary hypertension
- Portal vein thrombosis
Definition of decompensated cirrhosis:
- Decompensation is defined as the development of “major” cirrhosis complications which include portal hypertensive GI bleeding, new onset of ascites, SBP, HE, HRS, hepatopulmonary syndrome, and hepatocellular carcinoma *.
- Other complications of cirrhosis include portal vein thrombosis and portopulmonary hypertension. However, patients with these complications alone are not considered to have decompensated cirrhosis.
- Likewise, patients with varices but who have not developed variceal bleeding are not considered to have decompensated cirrhosis.
Clinical manifestation of decompensated cirrhosis
- Compensated cirrhosis is often asymptomatic. Patients have a median survival of more than 12 years *.
- In some patients with compensated cirrhosis, there’s no obvious abnormality on hepatic ultrasonography or liver function tests.
- Decompensated cirrhosis is marked by the development of “overt” clinical signs and symptoms, the most frequent of which are ascites, GI bleeding, delirium, jaundice, and infections.
- Once decompensation has occurred, cirrhosis becomes a systemic disease, with multi-organ/system dysfunction with a median survival of ~ 2 years *.
-
Patients with decompensated cirrhosis are highly susceptible to bacterial infections because of complex cirrhosis-associated immune dysfunction, which involves both innate and acquired immunity. In turn, patients with bacterial infections are burdened by severe morbidity, up to ACLF, and high mortality.
Pathophysiology of decompensation
- The pathogenesis of ascites and related cirrhosis complications are shown below.
- Cirrhosis causes systemic vasodilation due to a release of vasodilators (e.g. nitric oxide) from hepatic circulation.
- This causes a marked reduction of effective arterial blood volume and relatively insufficient cardiac output (and low blood pressure), which in turn kicks off activation of the renin-angiotensin-aldosterone system, sympathetic nervous system, and arginine-vasopressin secretion (releasing angiotensin II, norepinephrine and ADH).
- Initially, these systems can correct systemic vasodilation and hypotension. Beyond a certain point, these are inefficient, and refractory systemic vasodilation ensues. This is clinically marked by hypotension and multi-organ/system hypoperfusion and dysfunction.
- Vasodilation and increased permeability of the bowel capillaries lead to bacterial translocation and endotoxemia. Chronic exposure to sublethal levels of endotoxin will prime endothelial cells to overproduction of nitric oxide, which further perpetuates systemic vasodilation and organ hypoperfusion *.

- In ~ 40-50% of patients with decompensated cirrhosis, no identifiable precipitating factor can be identified.
- Possible precipitating factors include *:
- Infection (including spontaneous bacterial peritonitis, urosepsis, pneumonia, Clostridioides difficile infection). Is the most common cause of decompensation of cirrhosis.
- Hemorrhage (e.g. variceal bleeding, portal gastropathy, peptic ulceration)
- Thrombosis (e.g. portal vein thrombosis, hepatic vein thrombosis a.k.a. Budd-Chiari syndrome)
- Hepatic insult:
- Alcoholic hepatitis
- Drug-induced liver injury (especially acetaminophen toxicity).
- Biliary obstruction.
- Viral infection: acquisition of new viral infection or flare of chronic viral hepatitis.
- Hemodynamic abnormalities:
- Hypovolemia (e.g. due to excess diuretic, poor PO intake, Large volume paracentesis without albumin replacement).
- Hypervolemia (e.g. due to cirrhosis with volume retention).
- Medications (e.g. antihypertensives, vasodilators, nephrotoxins).
Evaluation
- Patients with decompensated cirrhosis should have thorough investigations to assess the severity of the disease, complications, and etiology of the underlying cirrhosis (above).
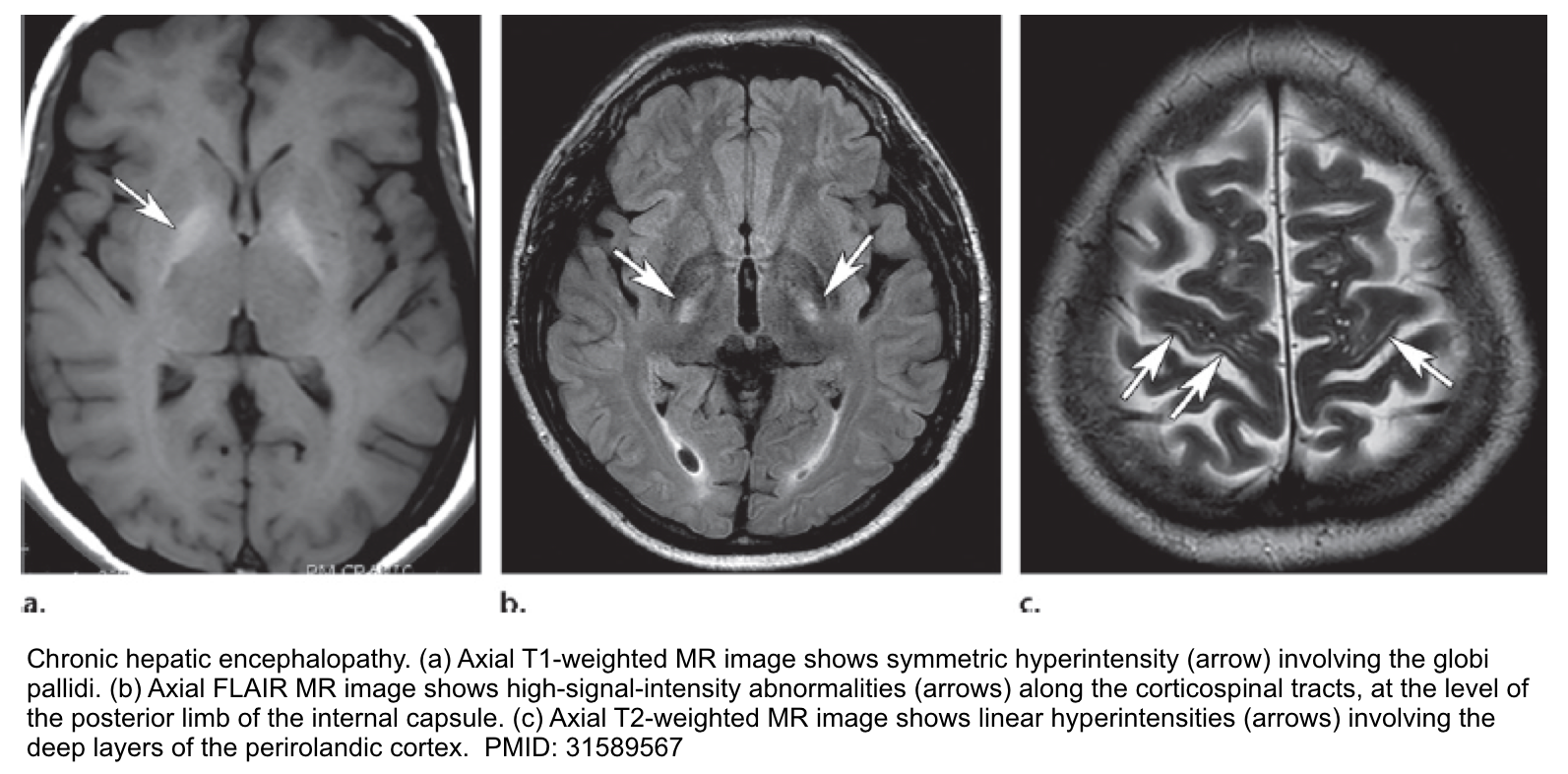
- In delirious patients consider brain CT/MRI to evaluate for other etiologies of neurologic dysfunction. Brain MRI findings in patients with chronic hepatic encephalopathy may include:
- Bilateral, symmetric T1 hyperintensity in the globus pallidum and substantia nigra *.
- This is commonly seen among patients with chronic hepatic failure. Manganese accumulation in the brain seems to be the cause due to portosystemic shunting that bypasses liver uptake of manganese.
- Vasogenic edema may be seen involving the cerebral white matter (especially the corticospinal tract). Although cerebral edema is more typically associated with acute liver failure, this may even occur in chronic hepatic encephalopathy *.
- Hepatic encephalopathy is a common cause of acute toxic leukoencephalopathy.
- Laminar T2 hyperintensities may be seen involving the deep layers of the perirolandic cortex (the cortex near the central fissure)*.
- Bilateral, symmetric T1 hyperintensity in the globus pallidum and substantia nigra *.
- In delirious patients consider brain CT/MRI to evaluate for other etiologies of neurologic dysfunction. Brain MRI findings in patients with chronic hepatic encephalopathy may include:
Principles of management
The overall schema for the management of patients with decompensated cirrhosis includes *:
- Preventing further insult to the liver; for example avoidance of hepatotoxins and medication adjustment.
- Specific therapies are directed against the underlying cause of cirrhosis, for example, antiviral therapy for HBV, and abstinence from alcohol in alcoholic cirrhosis (Table above).
- Treatment of the “current” complications
- Strategies to prevent the development of complications, for example, prophylactic non-selective beta blocker or endoscopic variceal ligation for those varices with increased risk of bleeding (appendix)
- Determining the optimal timing for liver transplantation (once decompensation has developed, patients should be considered for liver transplantation).
Hepatic encephalopathy in patients with cirrhosis
- HE is divided into 3 types based on the underlying liver disease *
- Type A: resulting from acute liver disease (see above)
- Type B: resulting from portosystemic shunting without any liver disease.
- Type C: resulting from cirrhosis. This is discussed here.
- Diagnosis of HE is made clinically and after excluding other causes of delirium. Virtually, there’s no sensitive and specific diagnostic test.
- Differential diagnosis of HE is mentioned above. Differentiating decompensated cirrhosis from HE might be challenging with the following conditions:
- Neurologic Wilson disease
- Features that suggest neurologic Wilson disease rather than hepatic encephalopathy include the presence of dysarthria, dystonia, tremors, or parkinsonism. In addition, neurologic symptoms in a patient with Wilson disease may have been present before the onset of the hepatic manifestations.
- Alcohol withdrawal
- In patients with alcoholism cirrhosis, differentiating hepatic encephalopathy from alcohol withdrawal may become challenging. However, these two are essentially opposite.
- Generally, this differentiation can be made based on history and physical examination. Features suggestive of alcohol withdrawal may include:
- Agitated delirium and seizure (vs. hypoactive delirium and coma in patients with HE).
- Tremor, hypertension, tachycardia, and visual hallucination (vs. asterixis in HE).
- The pathophysiology of alcohol withdrawal delirium involves inadequate excitation of GABA receptors with subsequent neuroexcitation. In contrast, patients with HE have excessive GABA-receptor stimulation by various toxins.
- Neurologic Wilson disease
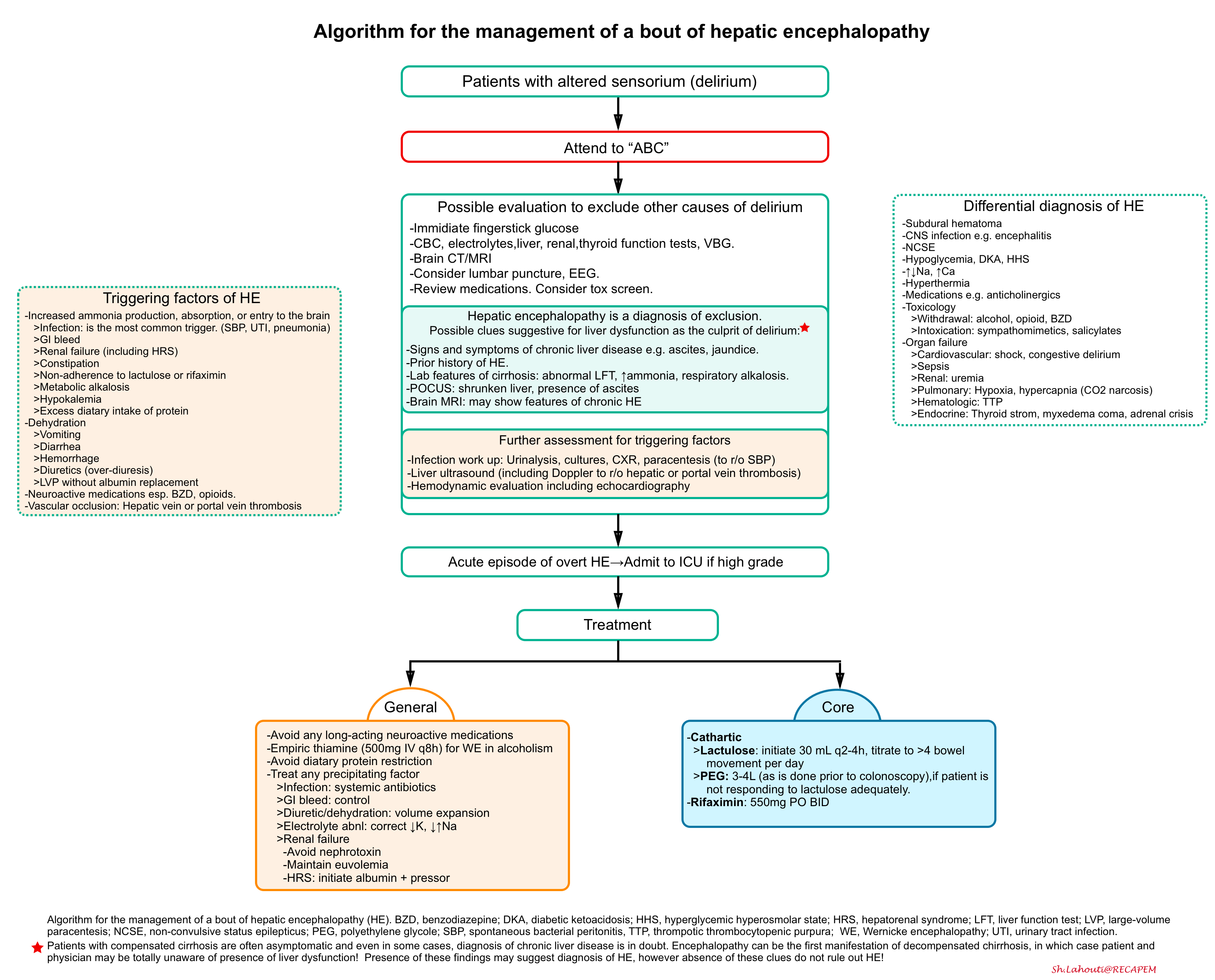
- The overall approach to diagnosis and management of HE resulting from cirrhosis is presented below *.
The principles of management
- Excluding other causes of altered sensorium by appropriate evaluation.
- Assessment for possible precipitating factors (e.g. infection, especially spontaneous bacterial peritonitis)
- Treatment plan
- The core treatment of HE involves measures to reduce ammonia production *:
- Cathartic
- Goal: To achieve >4 bowel movements per day.
- Lactulose. Start at a high dose and then titrate downwards (e.g. 30 ml q2h until frequent bowel movements, then decrease to 30 ml q6h and titrate).
- Polyethylene glycol (PEG)
- Recently multiple RCTs found that polyethylene glycol was superior to lactulose, in terms of achieving more rapid resolution of hepatic encephalopathy *.
- PEG may be used as add-on therapy for a patient who has been started on lactulose and isn’t responding adequately.
- Alternatively, in patients who develop hypernatremia as a side of lactulose treatment, transition to an isotonic solution of polyethylene glycol (e.g. GoLytely) should be considered.
- If PEG is used, it should initially be given with a large volume of water to avoid electrolyte shifts (using a standard 3-4 liter commercial preparation as is done before colonoscopy).
- Rifaximin
- This antibiotic is not absorbed through the gut and eradicates E. Coli which produces ammonia.
- Combination therapy involving rifaximin plus lactulose or PEG helps to reduce ammonia absorption from the bowel and has been shown to reduce length-of-stay, compared to lactulose/PEG monotherapy *.
- The usual dose is 400-550 mg PO BID.
- Cathartic
- The general treatment includes measures that remove coexisting or triggering factors *
- Avoid long-acting analgesics and opioid medications.
- Correct electrolyte disturbances especially hypokalemia and hyponatremia.
- Identify and treat precipitating factors
- In patients with cirrhosis, hepatic encephalopathy is most commonly triggered by infection, particularly “spontaneous bacterial peritonitis”.
- Have a low threshold to obtain cultures, and start empiric antibiotics if infection is highly suspected.
- Improve kidney function
- Adequate renal function is essential to allow patients to clear toxins and emerge from encephalopathy
- Appropriately treat possible conditions that are contributing to renal dysfunction. For example, if hypovolemia is clearly present, provide volume expansion with a balanced crystalloid.
- Avoid nephrotoxins and maintain euvolemia.
- Aggressively treat HRS.
- Avoid protein restriction
- The core treatment of HE involves measures to reduce ammonia production *:
Acute-on-chronic liver failure (ACLF)
- Acute-on-chronic liver failure (ACLF): refers to the most severe subset of patients with acutely decompensated cirrhosis presenting with failure of ≥2 extrahepatic organs (e.g. renal failure, severe encephalopathy, respiratory failure, shock, or severe coagulation abnormalities) in combination with worsened hepatic function.
- Liver and renal failures are the most common organ failures, followed by coagulation, brain, circulatory, and respiratory failure *.
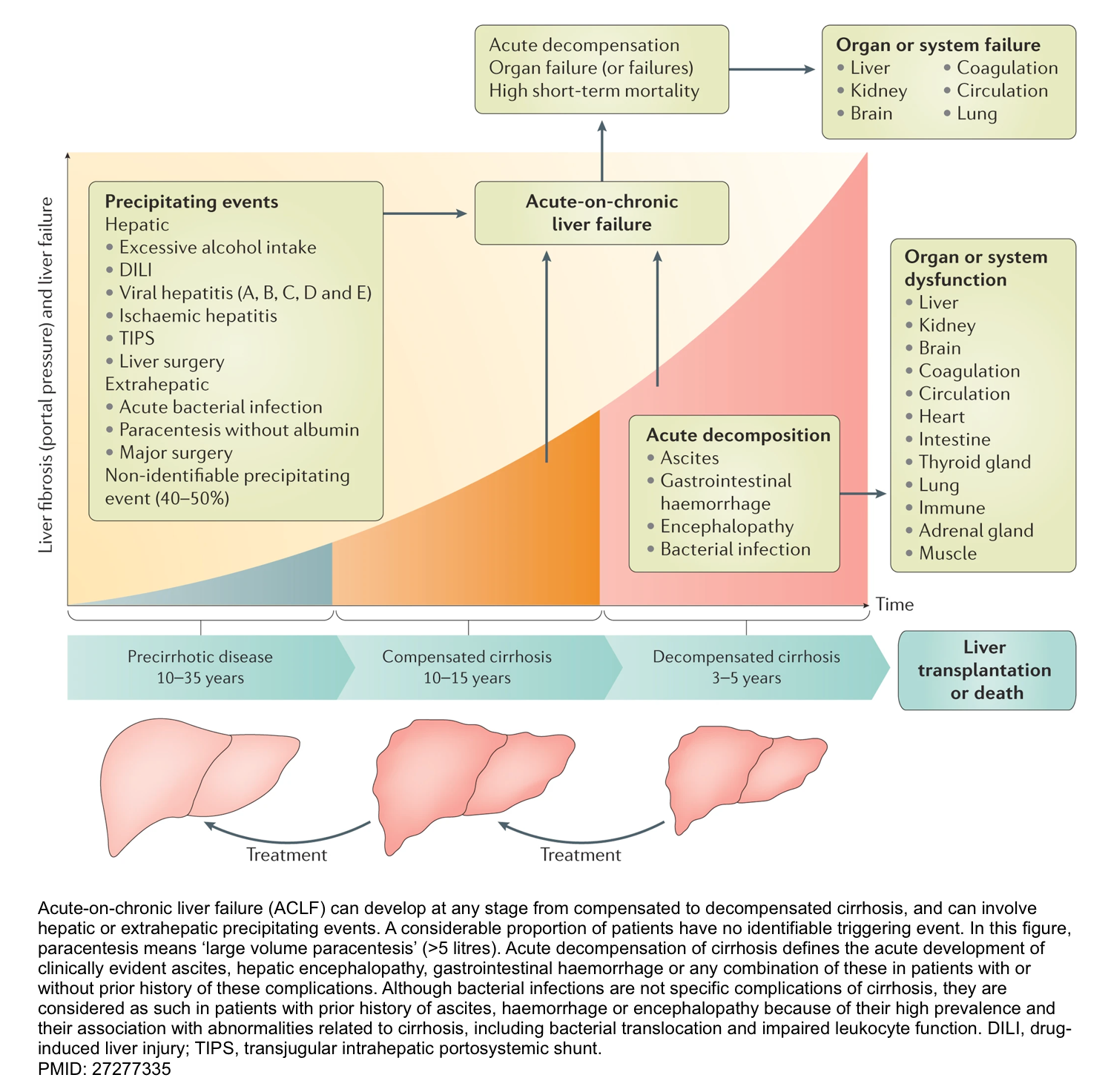
The pathophysiology of decompensated cirrhosis is discussed above. This emphasizes that patients with cirrhosis have systemic vasodilation and are vulnerable to any hemodynamic insults (i.e. precipitating factors), which if severe enough will cause profound vasodilation and multiorgan failure.
- The clinical course of cirrhosis is shown below. Acute‑on‑chronic liver failure (ACLF) can develop at any stage from compensated to decompensated cirrhosis and carries a high short-term mortality *.
Principles of evaluation and management
- The initial evaluation should be directed to identify possible trigger(s) of ACLF (explained above).
- If a cause of decompensation is discovered, it should be immediately treated (see specific treatment, table above).
- For example, prednisolone may be considered for some severe cases of alcoholic hepatitis (patients with Maddrey’s Discriminant Function >32).
- Management of ACLF is currently based on the supportive treatment of organ failures for all patients (whether or not a cause is found) shown below *.
- Consider candidacy for liver transplantation and discuss with a liver transplant service if this is a possibility.

Appendix

Going further
- Liver emergencies (emergency medicine cases)
- 5 Pearls on Hepatic Encephalopathy (Core IM Podcast)
- Coagulopathy in cirrhosis (IBCC)
Media
References
PMID: 27995906. Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am J Gastroenterol. 2017 Jan;112(1):18-35. doi: 10.1038/ajg.2016.517. Epub 2016 Dec 20.
PMID: 22213561. Lee WM, Stravitz RT, Larson AM. Introduction to the revised American Association for the Study of Liver Diseases Position Paper on acute liver failure 2011. Hepatology. 2012 Mar;55(3):965-7. doi: 10.1002/hep.25551.
PMID: 2841788. European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu; Clinical practice guidelines panel, Wendon, J; Panel members, Cordoba J, Dhawan A, Larsen FS, Manns M, Samuel D, Simpson KJ, Yaron I; EASL Governing Board representative, Bernardi M. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J Hepatol. 2017 May;66(5):1047-1081. doi: 10.1016/j.jhep.2016.12.003.
PMID: 28131021. Cardoso FS, Marcelino P, Bagulho L, Karvellas CJ. Acute liver failure: An up-to-date approach. J Crit Care. 2017 Jun;39:25-30. doi: 10.1016/j.jcrc.2017.01.003. Epub 2017 Jan 19.
PMID: 31840297. Dong V, Nanchal R, Karvellas CJ. Pathophysiology of Acute Liver Failure. Nutr Clin Pract. 2020 Feb;35(1):24-29. doi: 10.1002/ncp.10459. Epub 2019 Dec 15
PMID: 33838857. Rudler M, Weiss N, Bouzbib C, Thabut D. Diagnosis and Management of Hepatic Encephalopathy. Clin Liver Dis. 2021 May;25(2):393-417. doi: 10.1016/j.cld.2021.01.008. Epub 2021 Mar 11.
PMID: 27783916. Wijdicks EF. Hepatic Encephalopathy. N Engl J Med. 2016 Oct 27;375(17):1660-1670. doi: 10.1056/NEJMra1600561.
PMID: 22541696. Davern TJ. Drug-induced liver disease. Clin Liver Dis. 2012 May;16(2):231-45. doi: 10.1016/j.cld.2012.03.002.
PMID: 28346236. Lightsey JM, Rockey DC. Current concepts in ischemic hepatitis. Curr Opin Gastroenterol. 2017 May;33(3):158-163. doi: 10.1097/MOG.0000000000000355.
PMID: 24369077. Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013 Dec 26;369(26):2525-34. doi: 10.1056/NEJMra1208937.
PMID: 25117134. Ge PS, Runyon BA. Serum ammonia level for the evaluation of hepatic encephalopathy. JAMA. 2014 Aug 13;312(6):643-4. doi: 10.1001/jama.2014.2398
PMID: 2194520. Chavarria L, Alonso J, Rovira A, Córdoba J. Neuroimaging in acute liver failure. Neurochem Int. 2011 Dec;59(8):1175-80. doi: 10.1016/j.neuint.2011.09.003. Epub 2011 Sep 14.
PMID: 31589567. de Oliveira AM, Paulino MV, Vieira APF, McKinney AM, da Rocha AJ, Dos Santos GT, Leite CDC, Godoy LFS, Lucato LT. Imaging Patterns of Toxic and Metabolic Brain Disorders. Radiographics. 2019 Oct;39(6):1672-1695. doi: 10.1148/rg.2019190016
PMID: 1568412. Giannini EG, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005 Feb 1;172(3):367-79. doi: 10.1503/cmaj.1040752.
PMID: 11500606. Gill RQ, Sterling RK. Acute liver failure. J Clin Gastroenterol. 2001 Sep;33(3):191-8. doi: 10.1097/00004836-200109000-00005
PMID: 28131021. Cardoso FS, Marcelino P, Bagulho L, Karvellas CJ. Acute liver failure: An up-to-date approach. J Crit Care. 2017 Jun;39:25-30. doi: 10.1016/j.jcrc.2017.01.003. Epub 2017 Jan 19.
PMID: 30694840. Trovato FM, Rabinowich L, McPhail MJW. Update on the management of acute liver failure. Curr Opin Crit Care. 2019 Apr;25(2):157-164. doi: 10.1097/MCC.0000000000000583.
PMID: 28417882. European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu; Clinical practice guidelines panel, Wendon, J; Panel members, Cordoba J, Dhawan A, Larsen FS, Manns M, Samuel D, Simpson KJ, Yaron I; EASL Governing Board representative, Bernardi M. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J Hepatol. 2017 May;66(5):1047-1081. doi: 10.1016/j.jhep.2016.12.003.
PMID: 19394004. Amorós A, Aparicio JR, Garmendia M, Casellas JA, Martínez J, Jover R. Deep sedation with propofol does not precipitate hepatic encephalopathy during elective upper endoscopy. Gastrointest Endosc. 2009 Aug;70(2):262-8. doi: 10.1016/j.gie.2008.10.038. Epub 2009 Apr 25.
PMID: 28611340. Nabi T, Nabi S, Rafiq N, Shah A. Role of N-acetylcysteine treatment in non-acetaminophen-induced acute liver failure: A prospective study. Saudi J Gastroenterol. 2017 May-Jun;23(3):169-175. doi: 10.4103/1319-3767.207711. PMID: 28611340.
PMID: 19524577. Lee WM, Hynan LS, Rossaro L, Fontana RJ, Stravitz RT, Larson AM, Davern TJ 2nd, Murray NG, McCashland T, Reisch JS, Robuck PR; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009 Sep;137(3):856-64, 864.e1. doi: 10.1053/j.gastro.2009.06.006. Epub 2009 Jun 12. Erratum in: Gastroenterology. 2013 Sep;145(3):695. Dosage error in article text.
PMID: 9711192. Jones AL. Mechanism of action and value of N-acetylcysteine in the treatment of early and late acetaminophen poisoning: a critical review. J Toxicol Clin Toxicol. 1998;36(4):277-85. doi: 10.3109/15563659809028022.
PMID: 1904133. Harrison PM, Wendon JA, Gimson AE, Alexander GJ, Williams R. Improvement by acetylcysteine of hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med. 1991 Jun 27;324(26):1852-7. doi: 10.1056/NEJM199106273242604
PMID: 31915608. Seetharam A. Intensive Care Management of Acute Liver Failure: Considerations While Awaiting Liver Transplantation. J Clin Transl Hepatol. 2019 Dec 28;7(4):384-391. doi: 10.14218/JCTH.2019.00032. Epub 2019 Nov 13.
PMID: 24463187. Severs D, Hoorn EJ, Rookmaaker MB. A critical appraisal of intravenous fluids: from the physiological basis to clinical evidence. Nephrol Dial Transplant. 2015 Feb;30(2):178-87. doi: 10.1093/ndt/gfu005. Epub 2014 Jan 23.
PMID: 33205036. Zaccherini G, Weiss E, Moreau R. Acute-on-chronic liver failure: Definitions, pathophysiology and principles of treatment. JHEP Rep. 2020 Sep 2;3(1):100176. doi: 10.1016/j.jhepr.2020.100176
PMID: 33657293. China L, Freemantle N, Forrest E, Kallis Y, Ryder SD, Wright G, Portal AJ, Becares Salles N, Gilroy DW, O’Brien A; ATTIRE Trial Investigators. A Randomized Trial of Albumin Infusions in Hospitalized Patients with Cirrhosis. N Engl J Med. 2021 Mar 4;384(9):808-817. doi: 10.1056/NEJMoa2022166.
PMID: 17599633. Eefsen M, Dethloff T, Frederiksen HJ, Hauerberg J, Hansen BA, Larsen FS. Comparison of terlipressin and noradrenalin on cerebral perfusion, intracranial pressure and cerebral extracellular concentrations of lactate and pyruvate in patients with acute liver failure in need of inotropic support. J Hepatol. 2007 Sep;47(3):381-6. doi: 10.1016/j.jhep.2007.04.015. Epub 2007 May 30.
PMID: 12654129. Harry R, Auzinger G, Wendon J. The effects of supraphysiological doses of corticosteroids in hypotensive liver failure. Liver Int. 2003 Apr;23(2):71-7. doi: 10.1034/j.1600-0676.2003.00813.x.
PMID: 12143048. Harry R, Auzinger G, Wendon J. The clinical importance of adrenal insufficiency in acute hepatic dysfunction. Hepatology. 2002 Aug;36(2):395-402. doi: 10.1053/jhep.2002.34514
PMID: 21652548. O’Riordan A, Brummell Z, Sizer E, Auzinger G, Heaton N, O’Grady JG, Bernal W, Hendry BM, Wendon JA. Acute kidney injury in patients admitted to a liver intensive therapy unit with paracetamol-induced hepatotoxicity. Nephrol Dial Transplant. 2011 Nov;26(11):3501-8. doi: 10.1093/ndt/gfr050. Epub 2011 Jun 6.
PMID: 19001057. Leithead JA, Ferguson JW, Bates CM, Davidson JS, Lee A, Bathgate AJ, Hayes PC, Simpson KJ. The systemic inflammatory response syndrome is predictive of renal dysfunction in patients with non-paracetamol-induced acute liver failure. Gut. 2009 Mar;58(3):443-9. doi: 10.1136/gut.2008.154120. Epub 2008 Nov 10
PMID: 19343322. Karvellas CJ, Pink F, McPhail M, Cross T, Auzinger G, Bernal W, Sizer E, Kutsogiannis DJ, Eltringham I, Wendon JA. Predictors of bacteraemia and mortality in patients with acute liver failure. Intensive Care Med. 2009 Aug;35(8):1390-6. doi: 10.1007/s00134-009-1472-x. Epub 2009 Apr 3.
PMID: 22735303. Agarwal B, Wright G, Gatt A, Riddell A, Vemala V, Mallett S, Chowdary P, Davenport A, Jalan R, Burroughs A. Evaluation of coagulation abnormalities in acute liver failure. J Hepatol. 2012 Oct;57(4):780-6. doi: 10.1016/j.jhep.2012.06.020. Epub 2012 Jun 23.
PMID: 30060258. Intagliata NM, Argo CK, Stine JG, Lisman T, Caldwell SH, Violi F; faculty of the 7th International Coagulation in Liver Disease. Concepts and Controversies in Haemostasis and Thrombosis Associated with Liver Disease: Proceedings of the 7th International Coagulation in Liver Disease Conference. Thromb Haemost. 2018 Aug;118(8):1491-1506. doi: 10.1055/s-0038-1666861. Epub 2018 Jul 30.
PMID: 29194678. Stravitz RT, Ellerbe C, Durkalski V, Schilsky M, Fontana RJ, Peterseim C, Lee WM; Acute Liver Failure Study Group. Bleeding complications in acute liver failure. Hepatology. 2018 May;67(5):1931-1942. doi: 10.1002/hep.29694. Epub 2018 Mar 26.
PMID: 31915608. Seetharam A. Intensive Care Management of Acute Liver Failure: Considerations While Awaiting Liver Transplantation. J Clin Transl Hepatol. 2019 Dec 28;7(4):384-391. doi: 10.14218/JCTH.2019.00032. Epub 2019 Nov 13
PMID: 12548507. Shami VM, Caldwell SH, Hespenheide EE, Arseneau KO, Bickston SJ, Macik BG. Recombinant activated factor VII for coagulopathy in fulminant hepatic failure compared with conventional therapy. Liver Transpl. 2003 Feb;9(2):138-43. doi: 10.1053/jlts.2003.50017.
PMID: 30855324. Saner FH, Bezinover D. Assessment and management of coagulopathy in critically-ill patients with liver failure. Curr Opin Crit Care. 2019 Apr;25(2):179-186. doi: 10.1097/MCC.0000000000000591.
PMID: 30986390. O’Leary JG, Greenberg CS, Patton HM, Caldwell SH. AGA Clinical Practice Update: Coagulation in Cirrhosis. Gastroenterology. 2019 Jul;157(1):34-43.e1. doi: 10.1053/j.gastro.2019.03.070. Epub 2019 Apr 12.
PMID: 28537975. Abouelkassem E, Hasan S, Mazzeffi MA, Planinsic RM, Sakai T, Tanaka KA. Reduced Requirement for Prothrombin Complex Concentrate for the Restoration of Thrombin Generation in Plasma From Liver Transplant Recipients. Anesth Analg. 2017 Aug;125(2):609-615. doi: 10.1213/ANE.0000000000002106.
PMID: 32058375. Nanchal R, Subramanian R, Karvellas CJ, Hollenberg SM, Peppard WJ, Singbartl K, Truwit J, Al-Khafaji AH, Killian AJ, Alquraini M, Alshammari K, Alshamsi F, Belley-Cote E, Cartin-Ceba R, Dionne JC, Galusca DM, Huang DT, Hyzy RC, Junek M, Kandiah P, Kumar G, Morgan RL, Morris PE, Olson JC, Sieracki R, Steadman R, Taylor B, Alhazzani W. Guidelines for the Management of Adult Acute and Acute-on-Chronic Liver Failure in the ICU: Cardiovascular, Endocrine, Hematologic, Pulmonary and Renal Considerations: Executive Summary. Crit Care Med. 2020 Mar;48(3):415-419. doi: 10.1097/CCM.0000000000004193.
PMID: 30190075. Brown SA, Axenfeld E, Stonesifer EG, Hutson W, Hanish S, Raufman JP, Urrunaga NH. Current and prospective therapies for acute liver failure. Dis Mon. 2018 Dec;64(12):493-522. doi: 10.1016/j.disamonth.2018.04.002. Epub 2018 Sep 3.
PMID: 32334790. Rifaie N, Saner FH. Critical care management in patients with acute liver failure. Best Pract Res Clin Anaesthesiol. 2020 Mar;34(1):89-99. doi: 10.1016/j.bpa.2020.01.004. Epub 2020 Jan 23.
PMID: 32102926. Bernardi M, Angeli P, Claria J, Moreau R, Gines P, Jalan R, Caraceni P, Fernandez J, Gerbes AL, O’Brien AJ, Trebicka J, Thevenot T, Arroyo V. Albumin in decompensated cirrhosis: new concepts and perspectives. Gut. 2020 Jun;69(6):1127-1138. doi: 10.1136/gutjnl-2019-318843. Epub 2020 Feb 26.
PMID: 29653741. European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu; European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018 Aug;69(2):406-460. doi: 10.1016/j.jhep.2018.03.024. Epub 2018 Apr 10. Erratum in: J Hepatol. 2018 Nov;69(5):1207.
PMID: 32618647. Bajaj JS, Lauridsen M, Tapper EB, Duarte-Rojo A, Rahimi RS, Tandon P, Shawcross DL, Thabut D, Dhiman RK, Romero-Gomez M, Sharma BC, Montagnese S. Important Unresolved Questions in the Management of Hepatic Encephalopathy: An ISHEN Consensus. Am J Gastroenterol. 2020 Jul;115(7):989-1002. doi: 10.14309/ajg.0000000000000603.
PMID: 34006606. Hoilat GJ, Ayas MF, Hoilat JN, Abu-Zaid A, Durer C, Durer S, Adhami T, John S. Polyethylene glycol versus lactulose in the treatment of hepatic encephalopathy: a systematic review and meta-analysis. BMJ Open Gastroenterol. 2021 May;8(1):e000648. doi: 10.1136/bmjgast-2021-000648.
PMID: 24849268. Kimer N, Krag A, Møller S, Bendtsen F, Gluud LL. Systematic review with meta-analysis: the effects of rifaximin in hepatic encephalopathy. Aliment Pharmacol Ther. 2014 Jul;40(2):123-32. doi: 10.1111/apt.12803. Epub 2014 May 21.
PMID: 33205036. Zaccherini G, Weiss E, Moreau R. Acute-on-chronic liver failure: Definitions, pathophysiology and principles of treatment. JHEP Rep. 2020 Sep 2;3(1):100176. doi: 10.1016/j.jhepr.2020.100176.
PMID: 27277335. Arroyo V, Moreau R, Kamath PS, Jalan R, Ginès P, Nevens F, Fernández J, To U, García-Tsao G, Schnabl B. Acute-on-chronic liver failure in cirrhosis. Nat Rev Dis Primers. 2016 Jun 9;2:16041. doi: 10.1038/nrdp.2016.41.

Add comment