

June 13, 2022 by Shahriar Lahouti.
CONTENTS
- Definition
- KIDGO criteria for diagnosis and staging of AKI
- Limitation of defining AKI
- Risk factors
- Causes of AKI
- Evaluation to determine the cause of AKI
- Pathophysiology
- Complications
- Emergent management
- Specific conditions
- Media
- Going further
- Appendix
- References
Definition
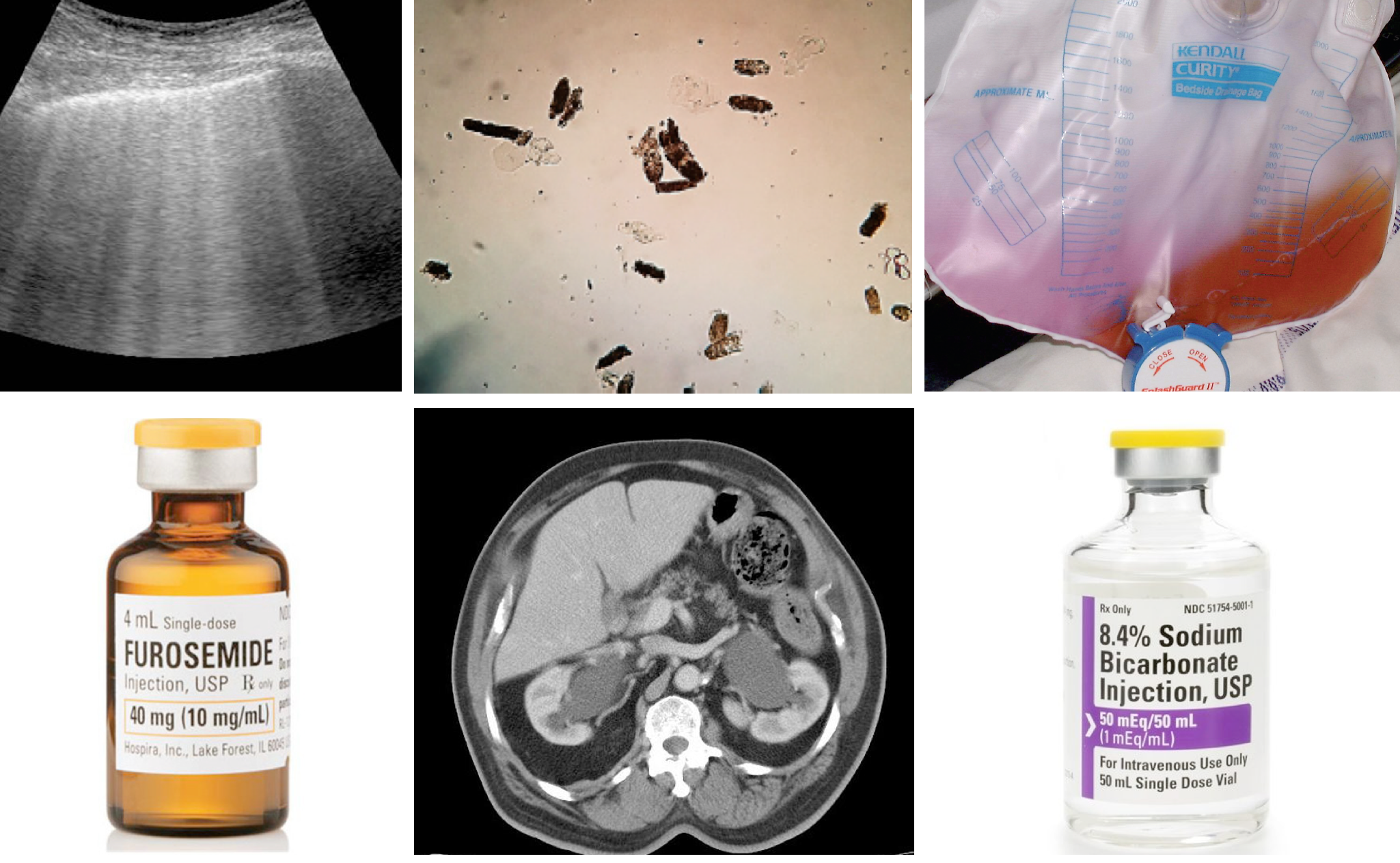
The KDIGO definition and staging system is the most recent and preferred definition. It defines AKI based on a rise in serum creatinine level (SC) compared to the baseline and/or fall in urine output (UO). The KDIGO guidelines define AKI by any of the following *:
- Increase in serum creatinine by ≥0.3 mg/dl within 48 hours, OR
- Increase in serum creatinine to ≥1.5 times baseline, which is known or presumed to have occurred within the prior seven days, OR
- Urine volume <0.5 mL/kg/hour for six hours.
KIDGO criteria for diagnosis and staging of AKI
This criteria allows for correction of volume status and obstructive causes of AKI prior to classification. Therefore before diagnosing and classifying AKI, one should assess and optimize volume status and exclude obstruction. Note that the basic assumption of these definition is that renal function has reached a steady state, which limits their applicability to ED patients with AKI whose renal function may be deteriorating.
- Staging of AKI
- As with other staging systems (e.g. RIFLE), the KDIGO suggested that patients can be classified (based on magnitude of “SC” rise or “UO” fall) to 3 stages of kidney injury with the highest stage correlating to a more severe injury and worse outcomes.
- Staging AKI based on these definitions has several caveats:
- These classification systems are independent of the various causes of AKI (prerenal, intrinsic, obstructive). While different causes of AKI are associated with different prognosis and grouping together these causes and treating AKI as a single disease is inappropriate.
- In a retrospective study of patients who developed AKI, in-hospital mortality and RRT rates shown to be dramatically different across each stage of AKI and is greatest when patients meet both the serum creatinine level and urine output criteria for AKI and also when these abnormalities persist (implying that the duration of AKI is also a significant predictor of important outcomes irrespective of severity) *.
Limitation of defining AKI
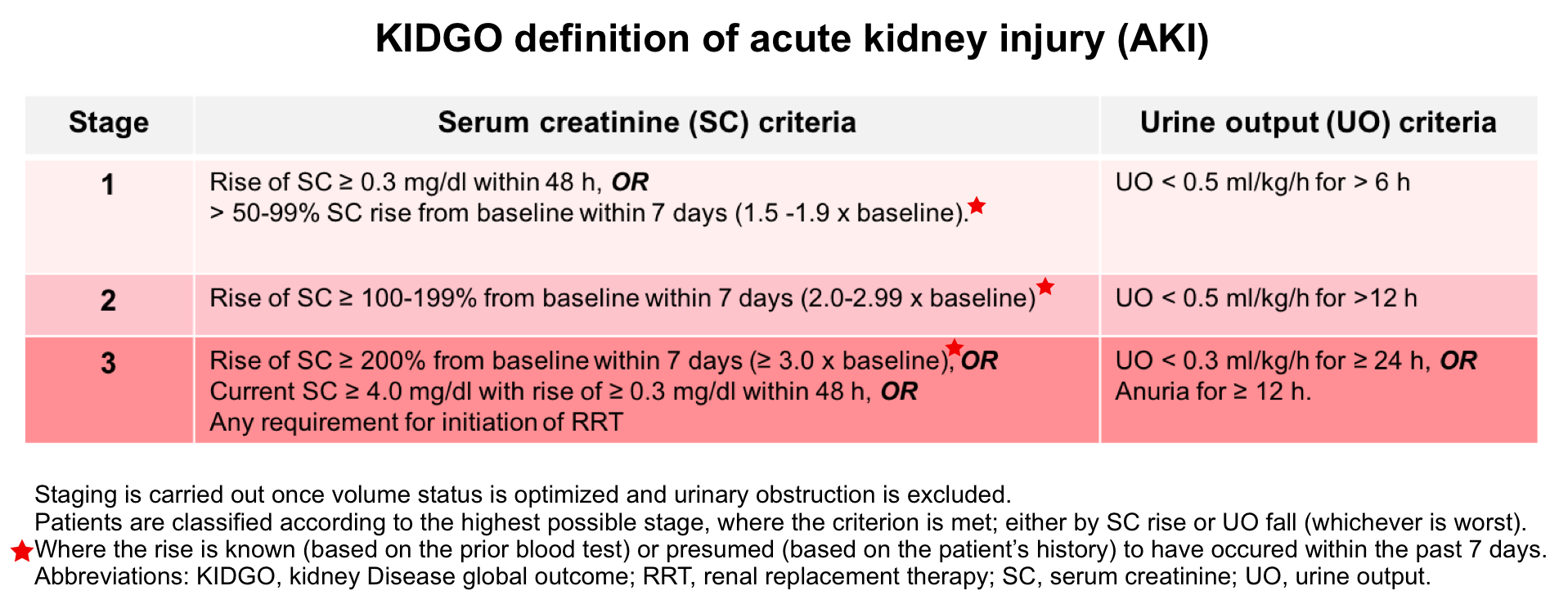
SC-based definition of AKI
- Physiologic background
- Serum creatinine is a metabolite of creatine and is synthesized from the amino acids glycine and arginine in liver, pancreas, and kidneys.
- Creatinine production is determined by the amount of creatine generated in liver, pancreas, and kidneys, creatine ingested (i.e. intake of red meat) and muscle mass and function *.
- In physiologic state, it is produced at a constant rate and the rate of production is matched by the rate of renal excretion. The half life of creatinine varies from 4 hours in healthy population to 24-72 hours in patients with kidney disease.
- Caveats
- Serum creatinine level is not an immediate marker of renal injury as the rise in serum level may lag 24-36h behind the onset of renal insult.
- In certain conditions such as sepsis, liver disease, serum creatinine production is declined, therefore the serum level does not provide accurate surrogate of kidney function * ,*.
- For example, a patient with cirrhosis and a creatinine of 1.0 mg/dL may have substantial renal dysfunction.
- Serum creatinine concentrations are also affected by drugs which compete with tubular secretion. In this case, serum creatinine levels may fluctuate without a change in renal function *.
- Determination of baseline creatinine: It is impossible to calculate the change in serum creatinine in patients who present with AKI but who do not have a baseline measurement of serum creatinine.
UO-based criteria for AKI
Definition
- By consensus, oliguria is defined as urine output < 0.5 ml/kg/h (roughly< 400ml/d) *.
- The occurrence of short periods (one to six hours) of oliguria lacked utility in discriminating patients with incipient AKI. More persistent oliguria (>12h) especially if intravascular volume is preserved is more worrisome.
Mechanism of oliguria
- Oliguria is a complex process and multiple mechanisms can potentially cause oliguria *.
- Oliguria is a physiologic response of functioning kidneys to prolonged fasting, hypovolemia. Here decrease in urine output is an adaptive response, in attempts to conserve volume.
- While traditional belief conceptualized oliguria as a surrogate for hypovolemia; this has been shown to be not true. In fact oliguria can be caused by any type of renal failure (if sufficiently severe). The mechanisms may include *
- Glomerular injury or altered intra-glomerular hemodynamics.
- Impaired tissue oxygenation causing preferential ischemia to the S3 segment of the proximal convoluted tubule and the thick ascending loop of Henle (since these parts of the nephron are more susceptible to ischemic lesions).
- Loss of osmolar gradient, interstitial edema, or inflammation.
- Lower urinary tract obstruction
Prognostic value
- Urine output is an important clinical marker but, like creatinine, is not renal specific. In fact, urine output may persist until renal function almost ceases.
- In critically ill patient, the onset of consistent oliguria is an ominous sign that requires immediate attention and intervention. In this context, oliguria is linked to important outcomes, such as AKI and increased morbidity and mortality *.
- Prognosis depends on change in urine output and serum creatinine level.
- Type A oliguria (oliguria with elevated serum creatinine, i.e. oliguric renal failure)
- It has been shown that patients meeting both urine output (UO) and serum creatinine (SC) criteria for AKI have dramatically worse outcomes compared with patients who manifest AKI solely or predominantly by one criterion.
- Type B oliguria (oliguria with stable serum creatinine level, aka. “isolated” oliguria)
- Isolated oliguria is a common finding in critically ill patients and is an independent risk factor for mortality *. Patients who meet stages 2 and 3 AKI by “UO criteria alone” have decreased 1-year survival (shown above).
- By enlarge, patients with isolated oliguria have a better prognosis relative to patients with type A oliguria.
- Type A oliguria (oliguria with elevated serum creatinine, i.e. oliguric renal failure)
Limitation of UO-based definition of AKI
- Several experts have questioned the validity of this arbitrary cut-off and suggest using either a longer minimum period (e.g. 12 h) or a lower threshold for urinary output (e.g. 0.3 ml/kg/h instead of 0.5 ml/kg/h) to reach sufficient specificity for diagnosing AKI *,*.
Risk factors
AKI can develop de novo in the setting of intact kidney function or can be superimposed on underlying chronic kidney disease (acute on chronic kidney injury). Risk factors for AKI include *
- Pre-existing CKD. This is the most important risk factor for AKI, and will ↑ the risk by as much as 10 fold compared to patients without CKD *,*.
- Nephrotoxins
- Hypovolemia
- Sepsis
- Diabetes
- Congestive heart failure
- Chronic liver disease
Causes of AKI
- Volume depletion (profuse diarrhea, vomiting, over-diuresis with diuretics)
- Shock of any etiology
- Cardiorenal syndrome
- Hepatorenal syndrome
- Congestive nephropathy (systemic congestion impairs venous outflow)
- Abdominal compartment syndrome
- Hypertensive emergency
- Thrombotic thrombocytopenic purpura & hemolytic uremic syndrome
- Tubular injury
- Ischemic ATN: Ischemia due to severe prolonged hypoperfusion (see pre-renal causes above).
- Nephrotoxic ATN:
- Endogenous toxins: Myoglobin (rhabdomyolysis), hemoglobin (hemolysis), intra-tubular obstruction from crystalluria (ethylene glycol, tumor lysis syndrome)
- Exogenous toxins: see the panel below
- Tubulointerstitial injury
- Acute allergic interstitial nephritis: NSAIDs, antibiotics
- Allograft rejection
- Infection: Viral, bacterial, and fungal infections
- Acute glomerulonephritis (1)
- Nephrolithiasis
- Prostate obstruction, e.g. BPH, carcinoma, infection.
- Bladder neck obstruction
- Occluded or mal-positioned Foley catheter
- Functional: neurogenic bladder secondary to spinal cord injury, diabetes, multiple sclerosis, stroke, pharmacologic side effects of drugs (anticholinergics, antidepressants).
- Urethral: posterior urethral valves, strictures, trauma.
- Nephrotic syndrome with peripheral edema, heavy proteinuria (protein excretion greater than 3.5 g/24h), hypoalbuminemia (<3g/dl), and hyperlipidemia and a bland urinary sediment.
- Nephritis syndrome with acutely elevated creatinine, hematuria, proteinuria, red blood cell casts, hypertension and no obvious cause of pre-renal or post-renal cause.
⚠️List of nephrotoxic drugs *
Antibiotics: Vancomycin, Aminoglycosides, Amphotericin, Sulfonamides, Pentamidine, Beta-lactams esp. nafcillin
Antivirals: Acyclovir, ganciclovir, valacyclovir, valganciclovir, foscarnet, Indinavir, cidofovir, tenofovir.
ACE-inhibitors, Angiotensin receptor blockers (ARBs).
NSAIDs.
Calcineurin inhibitors (cyclosporine, tacrolimus, sirolimus).
Chemotherapeutics (carboplatin, cisplatin, ifosfamide, methotrexate, mitomycin).
Mannitol.
Intravenous immunoglobulin (IVIG).
Inflammatory bowel disease medications (mesalamine, sulfasalazine).
Bisphosphonates (pamidronate, zoledronic acid).
Antiepileptics (topiramate, zonisamide).
Sodium chloride (0.9% or 3% in large quantity).
Synthetic cannabinoids
Poison
Oral phosphate bowel preparations
Evaluation to determine the “cause” of AKI
The assessment of the patient with AKI requires a meticulous history and physical examination, comprehensive review of medical records, evaluation of urinary findings, including the urinary sediment, review of laboratory tests, renal imaging. Although several mechanism might be involved in the development of AKI, some clinical and historical findings may be useful to sort out the cause of AKI.
Historical clues
- Volume loss, decreased cardiac output, suggest pre-renal cuases of AKI.
- Use of nephrotoxins, rhabdomyolysis, pulmonary renal diseases are suggestive for intrinsic renal disease.
- Alternating oliguria and polyuria is suggestive for obstructive process.
- Suspect postrenal AKI in men with prostatic disease or advanced age and patients with indwelling bladder catheters.
- Anuria strongly suggests obstruction, although vascular obstruction and fulminant renal disease are also possible.
Physical
- Distended palpable bladder suggests post-renal causes.
- Hypotension may suggest pre-renal failure.
- Hypertension (moderate-severe) is more suggestive of intrinsic renal failure (rather than pre-renal etiologies).
Labs
- Serum creatinine (attempt to ascertain a baseline).
- The serum creatinine tends to rise progressively and usually at a daily rate of ~1mg/dl/d with complete cessation of renal function.
- A higher rate of rise in serum creatinine (~>1.5-2 mg/dl per day) is highly suggestive for rhabdomyolysis.
- A slower rate of rise with periodic downward fluctuation (due to variation in renal perfusion) is suggestive of prerenal disease.
- Monitor urine output ideally with a urethral catheter
- Exact measurements of urine output require a urinary catheter to be in place for greater than 6h to meet the criteria of AK, limiting the applicability of urine output in an ED setting.
- Presence of oliguria is not helpful diagnostically; however absence of oliguria argues against a prerenal cause of AKI.
- Urinalysis. It can provide helpful information *,*. The interpretation in the context of AKI is summarized below.
- Creatinine Kinase (CK). Consider to request for CK when:
- Any one risk factor for rhabdomyolysis is present including trauma, compartment syndrome, crush injury, extreme exertion, hyperthermia, found down.
- Presence of any one symptom of rhabdomyolysis (more on this below) such as muscle pain, muscle weakness, vomiting, dark urine.
- Electrolytes. see below.
Pearls and pitfalls
- BCR (BUN/Cr ratio) >1 is not reliable at distinguishing pre-renal from renal causes *.
- Urine electrolytes & fractional excretion of sodium (FENa) previously were used to differentiate between “prerenal” and “intrinsic renal” failure. However, more recent evidence shows that FENa performs poorly *.
- Urine eosinophils has poor sensitivity and specificity for diagnosing acute tubulointerstitial nephritis.
- In a series of 566 patients who had urinary eosinophil testing and renal histology from kidney biopsy, eosinophiluria only had a 31% sensitivity and 68% specificity for the diagnosis of interstitial nephritis *.
- Furosemide stress test (FST) can be used in patients with AKI (stage ≤2) who has been appropriately resuscitated and obstructive causes have been excluded. This determines diuretic responsiveness and predicts worsening renal function. Insufficient urine production (< 200 mL over 2h) following administration of furosemide suggests intrinsic renal failure (see below).
Urinalysis interpretation in the context of AKI
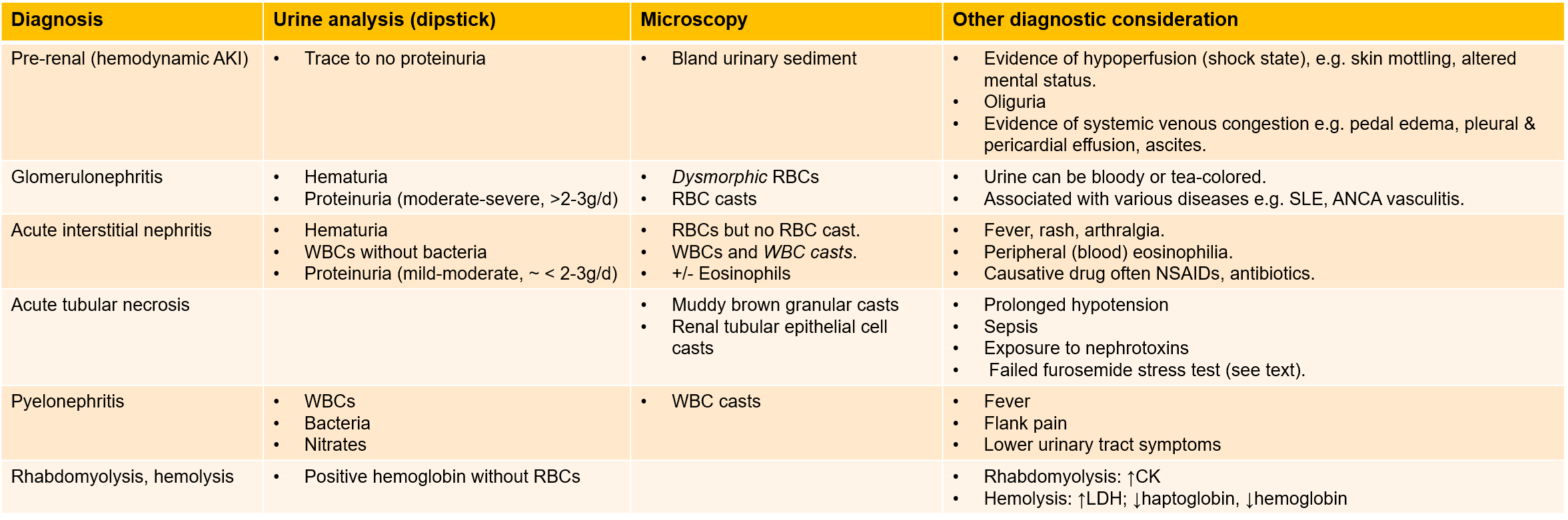
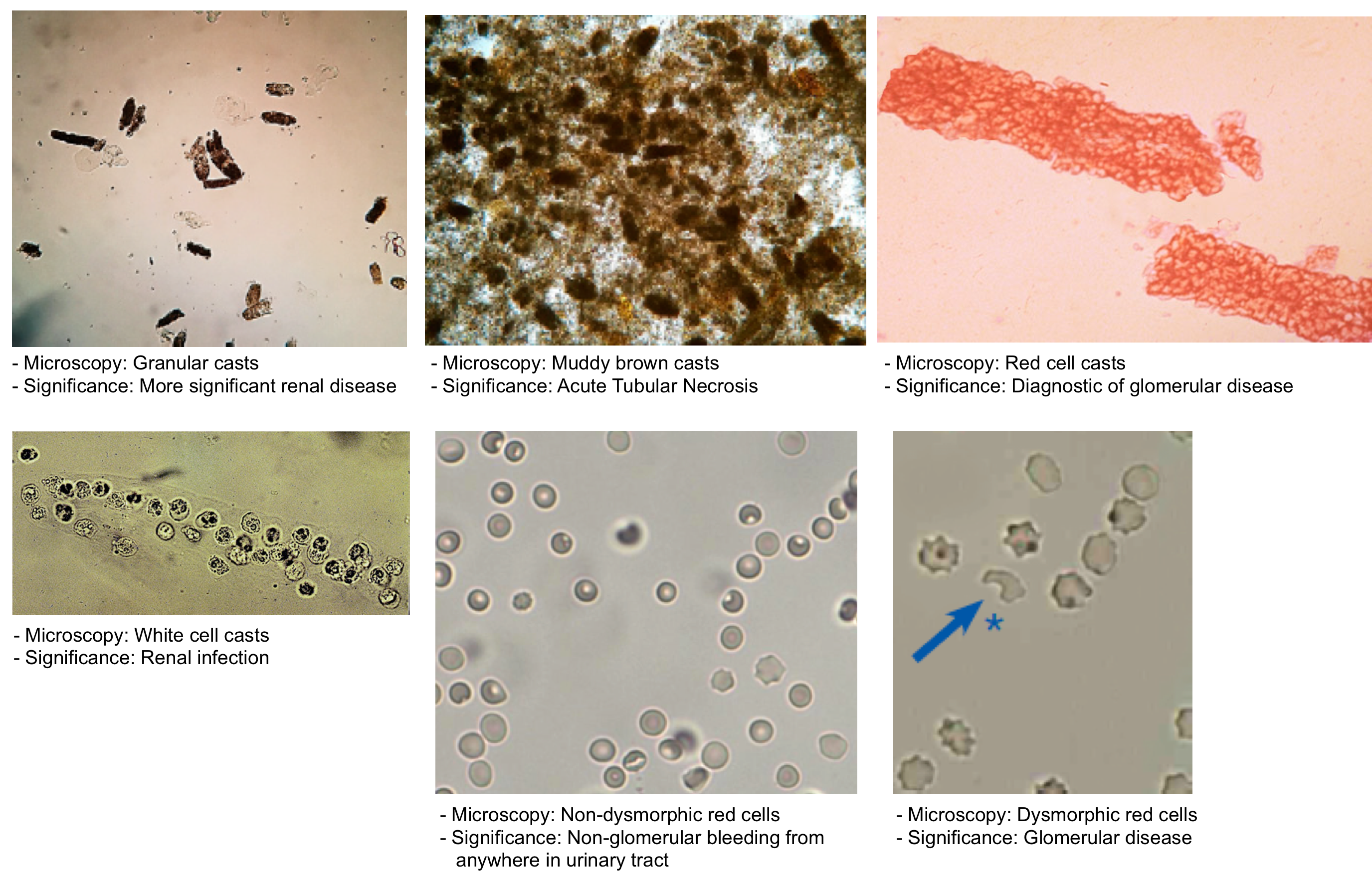
Imaging
Bedside ultrasound
- Urinary tract.
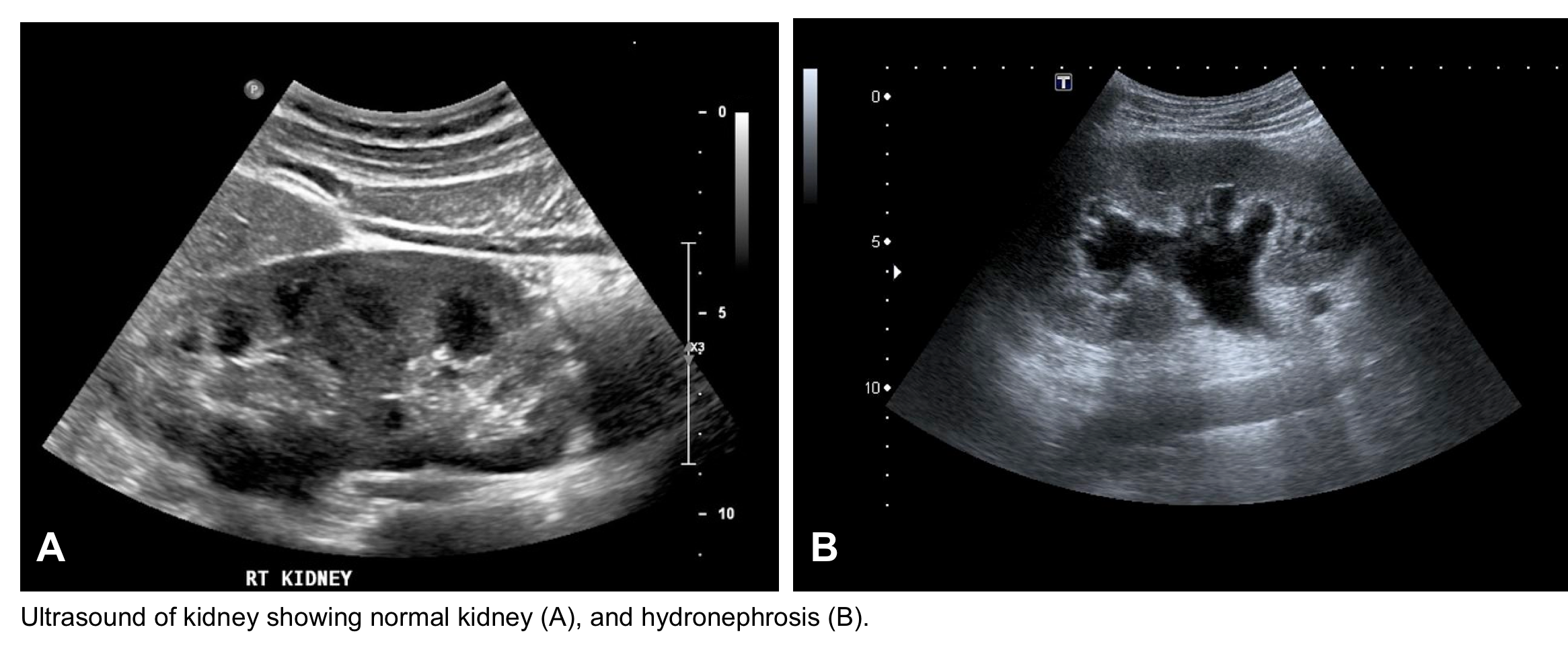
- Hydronephrosis and/or bladder distension. US of urinary tract is by enlarge most helpful for evaluating the obstructive causes of AKI.
- Hydronephrosis is a sensitive (98%) sign for urinary tract obstruction, however absence of hydronephrosis does not rule out obstruction especially in the volume-depleted patient, or in patients with obstruction caused by ureteric encasement or infiltration (e.g. retroperitoneal fibrosis, neoplasia). Furthermore, functional dilatation can occur in the setting of chronic ureteral reflux.
- Scan the bladder to assess urinary bladder volume. A postvoid bladder residual volume (PVR >~100-150 mL) suggests bladder outlet obstruction *. In patients with indwelling urinary catheter, a distended bladder indicates catheter malfunction (media 1).
- Renal size: Bipolar renal length is easy to assess by US, and kidney dimension of <9 cm suggests CKD.
- Renal parenchymal echogenicity: Renal parenchyma should be isoechoic or hypoechoic compared with that of the liver and spleen. Hyperechogenicity indicates diffuse parenchymal disease.
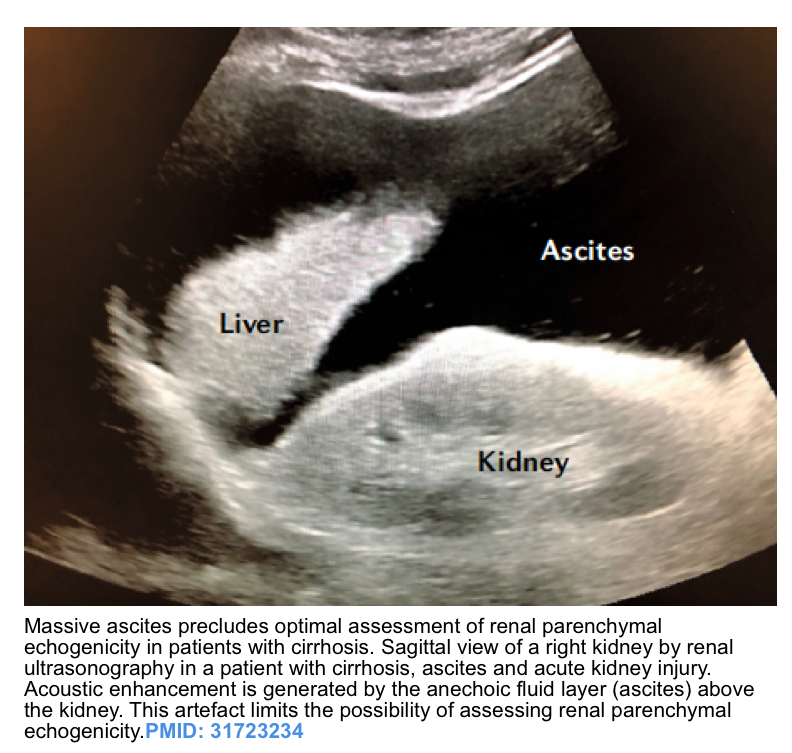
- Caveat: presence of massive ascites (as seen in HRS) precludes the optimal assessment of renal parenchymal echogenicity or corticomedullary differentiation due to the acoustic enhancement produced by the fluid layer (image below).
- More on renal ultrasound here.
- Hydronephrosis and/or bladder distension. US of urinary tract is by enlarge most helpful for evaluating the obstructive causes of AKI.
- Hemodynamic evaluation. POCUS to assess the heart, lung, IVC and Doppler of abdominal veins for assessing venous congestion (more on this here).
Non-contrast CT. If renal US detects hydronephrosis, a secondary imaging study (e.g. CT scan) to define the location of obstruction may be required.
Pathophysiology
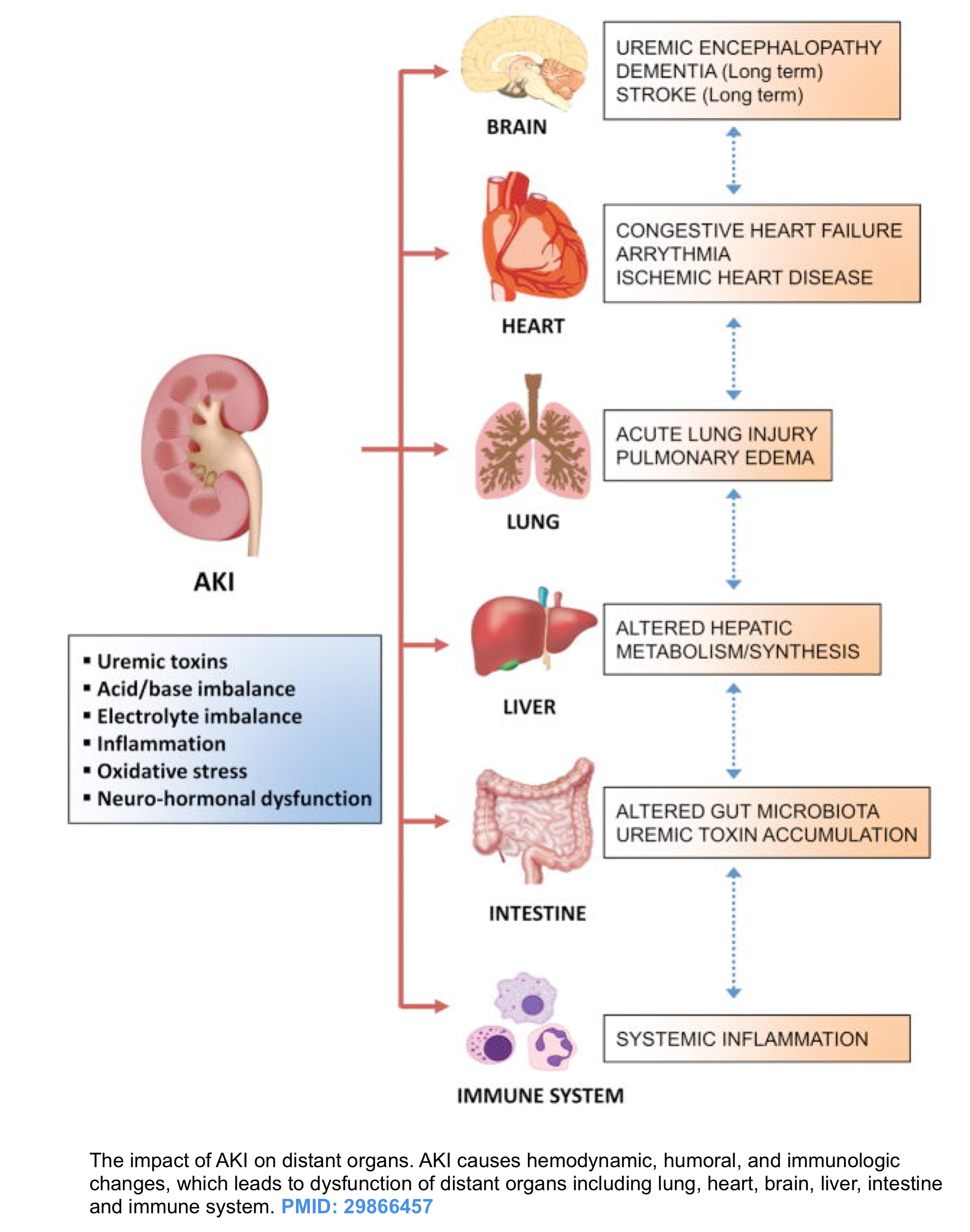
AKI can be viewed as a systemic disorder caused by an augmented inflammatory response and immunocompromise, volume overload and pulmonary edema, metabolic acidosis (reduced excretion of acid and reabsorption of bicarbonate), electrolyte abnormalities (potassium, calcium, phosphorus), GI edema, gut ischemia, and acute lung injury *, *.
Hypervolemia and fluid overload
Fluid overload is associated with increased morbidity and mortality *. The pathophysiology of hypervolemia and its deleterious impact on organ function discussed here. The vicious cycle of volume overload and kidney injury is illustrated below *,*.
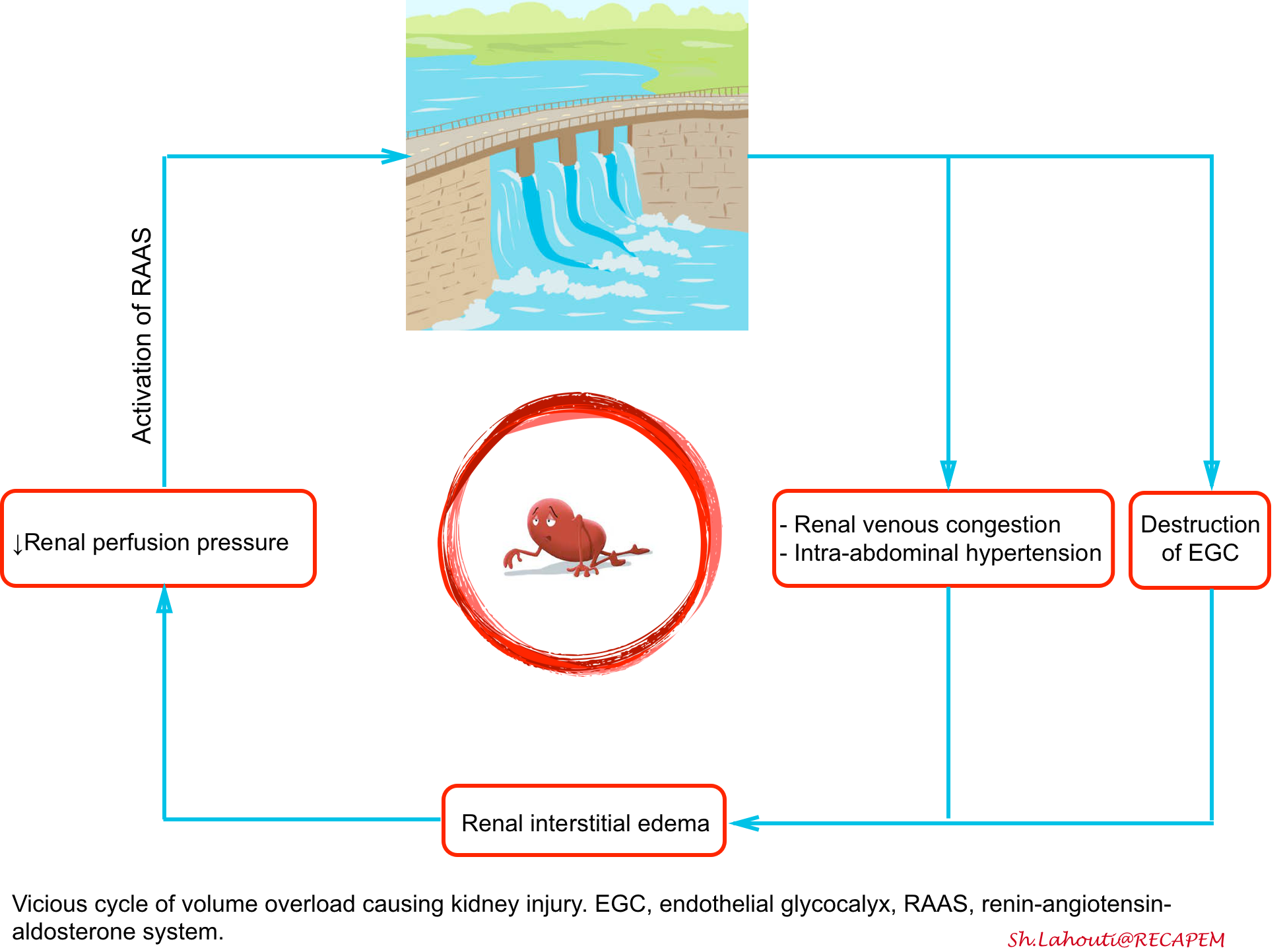
- Volume overload causes increased RV preload (↑CVP). This impairs renal perfusion by hindering venous blood flow out of the kidney (a.k.a. congestive nephropathy) *.
- Increasing venous pressure also produces renal interstitial edema. Because the kidney is enclosed by a capsule, interstitial edema increases subcapsular and intra-capsular pressure, leading to the reduction in forward renal arterial blood flow, reduction in venous return and lymphatic drainage, ultimately causing tissue hypoxia and AKI, analogous to compartment syndrome *.
- Moreover fluid overload is one of the major contributors to intra‐abdominal hypertension. This will further impair the kidney function (see below).
- Vascular congestion also destroy endothelial glycocalyx very rapidly. The resultant damage to the glycocalyx leads to increased vascular permeability, swelling of tissues, and worsening renal interstitial edema. This perpetuates renal hypoperfusion *.
- All mentioned mechanisms finally lead to renal hypoperfusion and decreased in afferent blood flow. This will activate the renin-angiotensin-aldosterone system (RAAS). Salt and water are thus retained as the kidney attempts to reverse what it senses as decreased effective volume, further exacerbating the fluid overload state *.
Kidney-lung interaction
- Acute lung injury is one of the most common extra-renal problems in AKI patients.
- The effects of AKI on lungs are largely due to pulmonary edema, which can be divided into *:
- Hydrostatic pulmonary edema which occurs mostly due to volume overload.
- Non-hydrostatic pulmonary edema (aka. nephrogenic pulmonary edema). This occurs without overt fluid overload as the integrity of alveolar-capillary barrier is impaired by uremia, systemic inflammation, and increased oxidative stress, causing fluid accumulation in the lung.
Complication of AKI
Metabolic acidosis
- AKI is commonly complicated by metabolic acidosis, typically with a widening of the serum anion gap due to retention of phosphates, sulfates, and organic anions.
👉Acidosis may be severe (daily fall in serum HCO3 >2 mmol/L) when the generation of H+ is increased by additional mechanisms (e.g. diabetic ketoacidosis, lactic acidosis).
Volume Overload and Cardiac Complications
- Extracellular volume overload is an almost inevitable consequence of diminished salt and water excretion in AKI and may present clinically as mild hypertension, ↑ central venous pressure, pleural effusion, ascites, pedal edema, ↑ body weight, and life-threatening pulmonary edema.
👉Moderate or severe hypertension is unusual in ATN and should suggest other diagnoses, such as hypertensive nephrosclerosis, glomerulonephritis, preeclampsia, renal artery stenosis, and other diseases of the renal vasculature.
Hematologic Complications
- Anemia develops rapidly in AKI and is usually multifactorial in origin. Contributing factors include inhibition of erythropoiesis, hemolysis, bleeding, hemodilution, and ↓RBC survival time.
- Prolongation of the bleeding time is also common, resulting from mild thrombocytopenia, platelet dysfunction, and clotting factor abnormalities (e.g. factor VIII dysfunction).
- ↑K
- It is a common and potentially life-threatening complication of AKI.
- The serum potassium level typically rises by 0.5 mmol/L/day in oligo-anuric patients; reflecting impaired excretion of potassium.
- 👉Hyperkalemia may be compounded by coexistent metabolic acidosis and/or hyperglycemia or other hyperosmolar states that promote potassium efflux from the cells.
- 👉Rapidly developing severe hyperkalemia at the time of diagnosis of AKI suggests massive tissue destruction, as might be seen with rhabdomyolysis, hemolysis, or tumor lysis syndrome.
- ↓K
- It is unusual in AKI but may complicate nonoliguric ATN caused by aminoglycosides, cisplatin, or amphotericin B, presumably because of impaired K reabsorption resulting from epithelial cell injury in the thick ascending limb of the loop of Henle.
- Hyperuricemia
- Uric acid is cleared from blood by glomerular filtration and secretion by proximal tubule cells.
- Asymptomatic hyperuricemia (12−15 mg/dl) is typical in established AKI.
- 👉Higher levels suggest increased production of uric acid and may point to a diagnosis of acute urate nephropathy.
- ↑PO4
- Mild to moderate hyperphosphatemia (5−10 mg/dl) is a common consequence of AKI.
- 👉Severe hyperphosphatemia (10−20 mg/dl) may be seen in highly catabolic patients or when AKI is associated with rapid cell death as in rhabdomyolysis, severe burns, hemolysis, or tumor lysis.
- ↓Ca
- Factors that potentially contribute to hypocalcemia include skeletal resistance to the actions of parathyroid hormone, reduced levels of 1,25-dihydroxyvitamin D, Ca sequestration in injured tissues, such as muscle in the setting of rhabdomyolysis, and metastatic deposition of calcium phosphate salts in the setting of severe hyperphosphatemia.
- Hypocalcemia is usually asymptomatic, possibly because of the counterbalancing effects of acidosis on neuromuscular excitability.
- 👉However, symptomatic hypocalcemia can occur in patients with rhabdomyolysis or acute pancreatitis or after treatment of acidosis with HCO3.
- ↑Mg
- Mild asymptomatic hypermagnesemia is common in oliguric AKI and reflects the impaired excretion of ingested magnesium, dietary magnesium, magnesium-containing laxatives, or antacids.
- More significant hypermagnesemia is usually the result of overzealous parenteral magnesium administration, as in the management of AKI associated with preeclampsia.
- ↓Mg occasionally complicates nonoliguric ATN associated with cisplatin or amphotericin B and, as with hypokalemia, likely reflects injury to the thick ascending limb of loop of Henle, a principal site for Mg reabsorption
Uremic syndrome
- Protracted periods of severe AKI or short intervals of catabolic anuric AKI often lead to the development of the uremic syndrome.
- Clinical manifestations of the uremic syndrome (in addition to those already listed above), include
- Anorexia, nausea, vomiting, metallic taste.
- Pericarditis, pericardial effusion, and cardiac tamponade.
- Gastrointestinal complications such as anorexia and ileus.
- Uremic bleeding. The major clinical manifestation of this is cutaneous bleeding, but GI bleeding can also occur.
- Uremic encephalopathy
- Impaired mental status e.g. lethargy, confusion, stupor, coma, agitation.
- Movement disorder: asterixis, tremor, myoclonus, hyperreflexia, restless legs syndrome.
- Seizures.
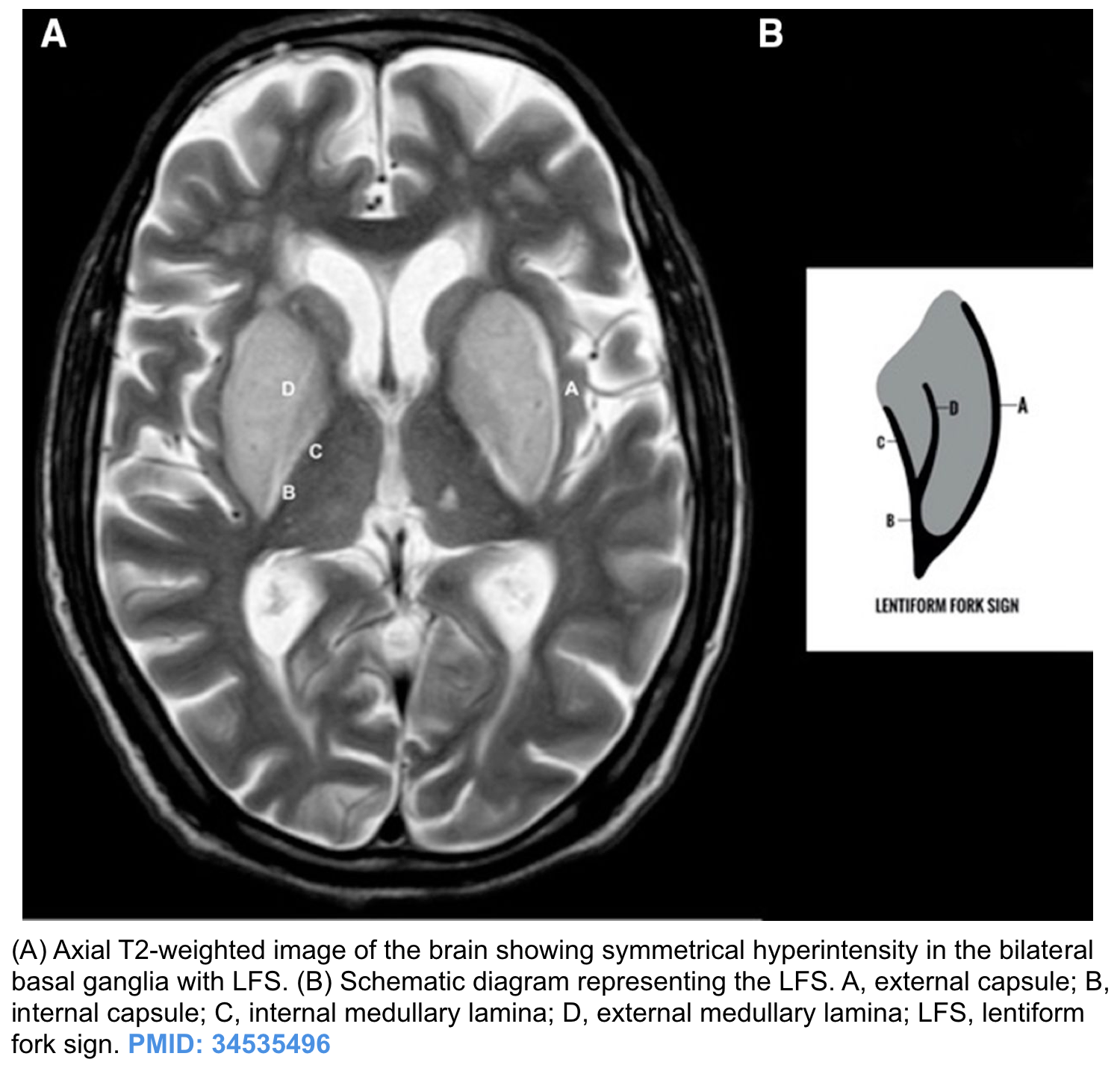
- Neuroimaging in uremic encephalopathy. Three patterns have been recognized *:
- Basal ganglia (bilateral symmetrical) involvement is the most common (figure below) pattern.
- Lentiform fork sign is a characteristic feature of uremic encephalopathy, in which the white matter surrounding the basal ganglia, the internal and external capsules and the medullary lamina become hyperintense on T2/FLAIR *.
- Cortical or subcortical involvement (PRES-like): Uremia may promote the development of posterior reversible encephalopathy syndrome (PRES), especially in combination with hypertension.
- Acute toxic leukoencephalopathy (ATL).
- Basal ganglia (bilateral symmetrical) involvement is the most common (figure below) pattern.
CKD. Transition from AKI to chronic kidney disease
- AKI is one of many initiating events that contribute to chronic, progressive kidney disease.
- Some patients with AKI fully recover kidney function, whereas in others, the development of CKD is accompanied by a progressive decline in kidney function, leading ultimately to end-stage kidney disease (ESKD) *.
- Incomplete repair following AKI contributes to the progression to CKD and ESKD.
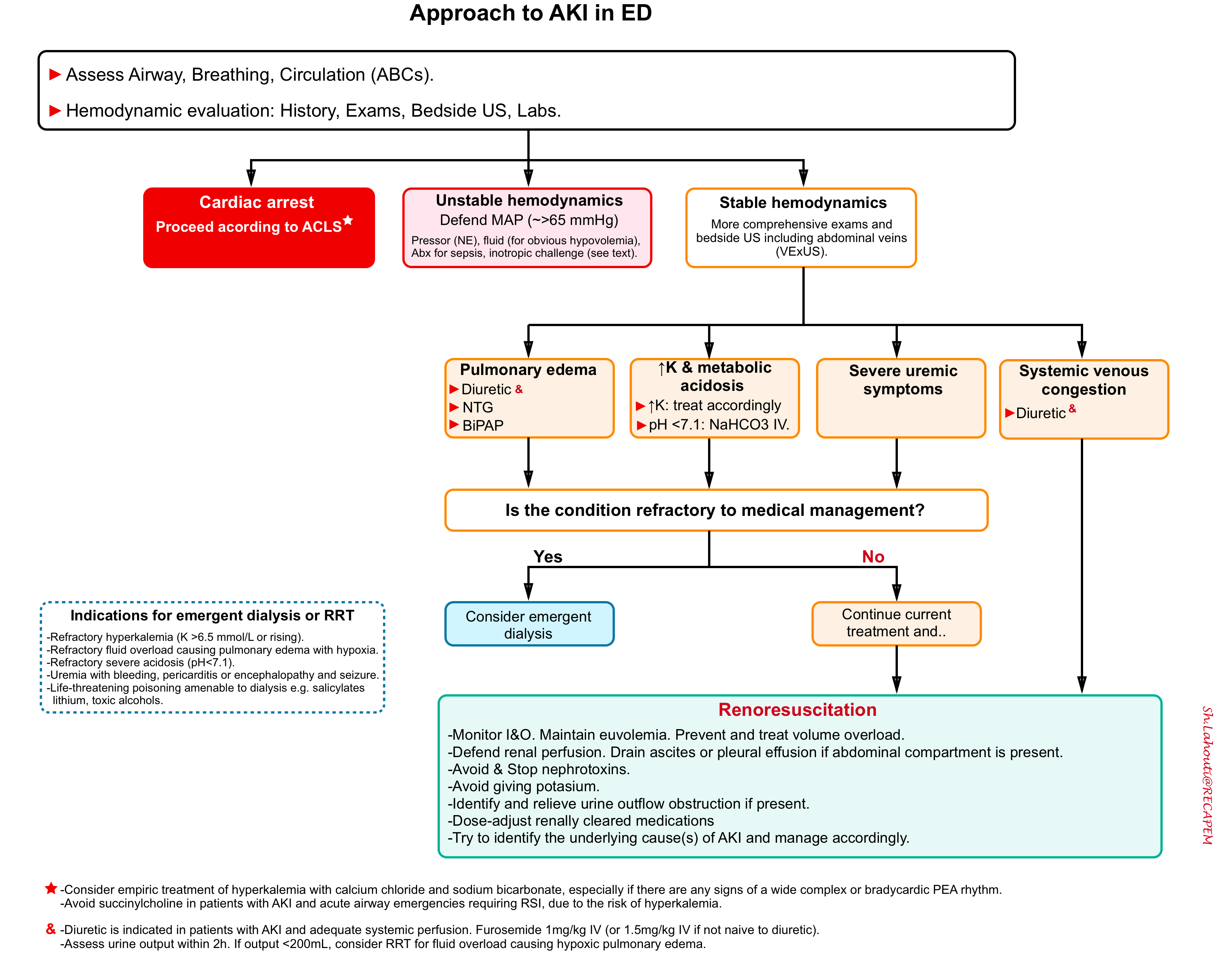
Emergent management of AKI in ED
For the critically ill patient with AKI, resuscitation is the first priority, and multiple diagnostic and therapeutic processes advance simultaneously. Generally, in the ED, the goals can be classified as:
- Identify and manage life-threatening causes of AKI (e.g. any type of shock).
- Optimize hemodynamics and defend renal perfusion (below).
- Identify and treat life-threatening presentations of AKI
- Renal Resuscitation.
- Monitor volume status. Maintain euvolemia and avoid volume overload.
- Optimize renal perfusion.
- Relieve urine outflow obstruction.
- Avoid/stop nephrotoxins. Avoid giving potassium.
- Dose-adjust renally cleared medications.
- Correct metabolic effects
The overall schema of management of AKI in ED is shown below.
Optimize kidney perfusion
Kidney perfusion pressure= MAP – CVP or IAP (whichever is higher)
Organ perfusion pressure is the difference between the inflow pressure (~MAP) and outflow pressure (~CVP).
- Inflow pressure. Although MAP is usually considered to be the inflow pressure, actual arterial inflow pressure varies greatly across the organs. Different pathologic conditions can also change the organ inflow pressure as well.
- For example, in patients with hypertension or CKD, the renal inflow pressure required to maintain adequate perfusion of the kidneys is higher than healthy individuals *.
- Outflow pressure. Despite that CVP is considered as outflow pressure, this is not uniform as well among different organs. For example in the presence of intra-abdominal hypertension or compartment syndrome, the global renal perfusion pressure is the difference between MAP and intra-abdominal pressure (IAP); when IAP exceeds CVP *.
Bottom line: An important aspect of renal resuscitation is to prevent and address ischemic insult by maintaining global renal perfusion pressure. The arterial side (maintaining MAP) has been focus of attention in the past, however as explored above; lowering venous pressure and/or intra-abdominal pressure are of paramount importance to keep global renal perfusion pressure at optimal level *.
Defend mean arterial pressure (MAP)
Target MAP. For most AKI patients, target MAP of > 65 mm Hg. However there is no “one-size fits all” for targeting the MAP.
- A higher MAP (>85 mmHg) for patients with risk factors for AKI (e.g. elderly, patients with chronic hypertension, atherosclerosis, and CKD) may improve outcome *,*.
- A higher MAP is also recommended in patients with markedly elevated CVP (e.g. RV failure due to severe pulmonary hypertension) to maintain systemic (and renal) perfusion pressure. Target a MAP of > (60 mmHg+ CVP).
- In patients with intra-abdominal hypertension, further increase in MAP target may be necessary, depending on their IAP. Target a MAP of > (60 mmHg + abdominal compartment pressure).
Vasopressor challenge
- Norepinephrine is the preferred vasopressors. However this should be tailored to individuals’ hemodynamics features.
- Vasopressin. In patients with AKI and septic shock, there is mixed evidence for preferential use of vasopressin over norepinephrine as the initial vasopressor of choice. A slower progression toward renal failure in vasopressin group compared to norepinephrine group has been shown particularly among patients with tachycardia and systemic vasodilation *.
- Dopamine. Pressor dose of dopamine has been shown to be associated with adverse events *. Low (“renal”)-dose dopamine does not improve renal recovery or decrease mortality *.
Inotrope challenge
- If there is evidence of poor cardiac output and concern for cardiogenic shock, it may be reasonable to try out an inotrope.
- Improved urine output following inotrope initiation confirms a diagnosis of cardiogenic shock. In this case, continue to treat the cardiogenic shock.
Fluid challenge
When fluid administration is appropriate?
- Fluid should only be given for total-body hypovolemia; after hemodynamic evaluation. The best indications for presence of true hypovolemia may include presence of ultrasonographic signs of hypovolemia and any of the following:
- Net negative balance, evidenced by monitoring intake/output (I&O) over the past 24 hours.
- Clinical history of profuse vomiting, diarrhea, overdiuresis and poor oral intake.
- Choice of fluid
- The fluid of choice is not normal saline (more on this here) *, *.
- Isotonic bicarbonate (D5W with 150 mEq/L sodium bicarbonate) is preferred for hypovolemic patients with uremic metabolic acidosis or non-anion gap metabolic acidosis (NAGMA),
- Balanced crystalloid (e.g. Lactated Ringers or plasmalyte) is the fluid of choice for patients with hypovolemia and normal serum bicarbonate *.
- Albumin. Volume resuscitation with albumin (rather than crystalloid) is preferred for patients with advanced liver cirrhosis following massive paracentesis and patients with hepatorenal syndrome (see below).
- See appendix for comparison of IV fluids and alkalizing agents.
- More: Fluid selection & pH-guided fluid resuscitation (IBCC)
Pulmonary edema
Patients who present with AKI and who have severe hypoxia due to pulmonary edema often need RRT urgently. Some patients may respond quickly to loop diuretics and may therefore avoid the need for RRT.
- Diuretic: can be administered to patient with pulmonary edema and adequate perfusion or patients with isolated peripheral edema and volume overload.
- Give furosemide 1 mg/kg IV (or 1.5 mg/kg IV if on furosemide already).
- Assess the clinical response. Consider a urine output of >200 mL within 2h of the furosemide dose as adequate.
- Patients who do not respond adequately to diuretics should be prepared for RRT.
- NTG: In a hemodynamically stable patient, a nitroglycerin bolus followed by infusion drip may be considered as a bridge to RRT.
- Optimize respiratory status.
- Drain ascites and/or pleural effusion in critical patients esp. if abdominal compartment syndrome is present, or patient’s respiratory status is deteriorating.
- Positive Pressure ventilation with BiPAP may be a adequate alternative to intubation in some fluid overloaded patients.
Metabolic acidosis
It has been demonstrated that bicarbonate use in the ICU for treatment of uremic metabolic acidosis avoids dialysis *.
Indication. In the absence of potential indications for acute RRT (see below), bicarbonate can be used for patients with AKI and severe metabolic acidosis (pH ≤7.1). Physiologic studies have suggested that, severe acidemia may produce hemodynamic instability related to reduced left ventricular contractility, arrhythmias, arterial vasodilation, and impaired responsiveness to catecholamine vasopressors *, *.
- In patients with anuria or fluid overload, dialysis is preferred because safe effective use of sodium bicarbonate requires urine flow and ability to tolerate a fluid load.
Dosage
- From a practical clinical perspective, the dose of bicarbonate to be administered is determined empirically.
- Almost 50 mEq/mL of bicarbonate will approximately increase serum bicarbonate by 1.5 mEq/L in a 70 kg patient.
- The rate of bicarbonate administration is dependent on severity of acidosis and volume status.
- The exact target level isn’t clear, but shooting for a pH >7.2 is reasonable (this is roughly equivalent to a bicarbonate level >17 mEq/L) *.
- Initial replacement. It can be administered as:
- Hypertonic bicarbonate: Bicarbonate IV is generally available as 8.4% (50 mEq/50 mL) or 7.2% (44.6 mEq/mL) and used for more sever acidosis. These formulations are hypertonic thus will raise serum sodium concentration and cause movement of water from intracellular to extracellular space.
- (50 ml ampules of 1 mEq/ml bicarbonate) can be used especially for patients with hyponatremia.
- Ampules should be pushed slowly over 10 minutes each, to avoid rapid swings in pH.
- In patients with baseline sodium ~140 mEq/L, this can cause hypernatremia.
- Isotonic bicarbonate: is prepared by mixing 150 mEq (or 3 ampules of 8.4% bicarbonate) into 1L of 5% dextrose.
- The infusion rate depends on severity of acidosis and the ability of the patient to tolerate volume overload.
- Hypertonic bicarbonate: Bicarbonate IV is generally available as 8.4% (50 mEq/50 mL) or 7.2% (44.6 mEq/mL) and used for more sever acidosis. These formulations are hypertonic thus will raise serum sodium concentration and cause movement of water from intracellular to extracellular space.
- Subsequent bicarbonate replacement. It can be given over the next 24 hours with isotonic bicarbonate (the maximum rate of administration is 1 mEq/kg/h).
Bicarbonate tablets: KDIGO guidelines suggest oral replacement when plasma bicarbonate concentrations are < 22 mEq/L. In the context of AKI, oral sodium bicarbonate tablets can also be used in patients who are able to tolerate oral administration of medications and who have mild acidosis (bicarbonate >18 mEq/L).
- Each 650 mg tablet contains 7.6 mEq of sodium bicarbonate.
- Depending on the severity of metabolic acidosis, the initial dose ranges between 15.4 to 23.1 mEq/day in divided doses (e.g. 650 mg tablet 2 to 3 times daily); titrate to normal serum bicarbonate concentrations (e.g. 23 to 29 mEq/L).
Complications: Volume overload, ↑ serum Na, ↓ K and ↓Ca.
- Bicarbonate administration may cause a decrease in the level of ionized or free calcium because of a pH-dependent increase in the binding of calcium to albumin. This could cause or worsen the symptoms of hypocalcemia (e.g. tetany) since symptoms of hypocalcemia reflect from the ionized, not total, calcium.
Renoresuscitation
1.Monitor volume status. Maintain Euvolemia and Avoid Volume Overload *.
- The vicious cycle of fluid overload, perpetuating AKI discussed above. Fluid overload is nephrotoxic and is associated with increased mortality in patients with AKI *.
- Assess volume status. Perform Doppler evaluation of abdominal veins for venous congestion. This can point to renal congestion (intra-capsular tamponade) as the cause of renal hypoperfusion by demonstrating the effects of raised RAP on venous outflow (VExUS, more on this here).
- Rememebr that oliguria is not a surrogate for hypovolemia, rather it can be caused by any type of renal failure. Blind administration of fluid to an oliguric patient is not necessarily the best move. Evaluate volume status before giving fluid.
- Goal: Target your resuscitation to achieve euvolemia.
- Volume overload
- Preventive strategy
- Therapeutic (decongestive) strategy.
- Diuretics are the mainstay of treatment for fluid overload.
- Furosemide stress test can be performed to assess for diuretic responsiveness.
- This a validated test of renal function to determine diuretic responsiveness as well as to predict worsening renal function. Furthermore it can guide the resuscitative efforts in oliguric patients (more on this here).
- Administer 1 mg/kg of furosemide in diuretic-naïve patients or 1.5 mg/kg in those with prior exposure. Measure urine output within 2h *.
- Preventive strategy
2.Optimize renal perfusion
- The principles of renal perfusion is explained above.
- Titrate vasopressor (if initiated) to keep MAP > ~65 mmHg.
- Relieve venous congestion (judicious diuretic use may be warranted in certain conditions).
- Drain massive ascites esp. if associated with abdominal compartment syndrome (more on this below).
3.Relieve urine outflow obstruction
- Timely relief of obstruction is essential for the return of normal renal function. Permanent loss of renal function develops over the course of 10 to 14 days in the setting of complete obstruction.
- Obtain bedside US to assess bladder volume. A PVR volume >125 mL suggests bladder outlet obstruction, for which a urethral catheter is placed.
4.Avoid nephrotoxins. In patients with AKI, stop medications such as NSAIDs, ACE inhibitors, ARBs and other nephrotoxins (see above), at least in the acute phase of AKI.
- Intravenous contrast media and AKI *, *
- The risk of contrast induced-AKI (CI-AKI) from intravenous iodinated contrast media is lower than previously thought *.
-
Contrast should never be withheld regardless of renal function if needed for life threatening diagnosis (e.g. aortic dissection, major trauma, etc.*).
-
The presence of a solitary kidney should not independently influence decision making regarding the risk of CI-AKI.
-
Patients receiving dialysis should be treated the same as those not receiving RRT; their dialysis schedule does not need to change.
5.Dose-adjust renally cleared medications
- Drugs that are renally cleared and that can produce serious adverse effects with accumulation in the setting of AKI should also be discontinued or undergo dose adjustment, even though they may not directly impact kidney function (e.g. morphine, metformin, gabapentin, cefepime).
- Dosing:
- Formulas for the GFR only work in steady state (equilibrium) conditions. This usually isn’t the case in acute kidney injury. Instead, the trend in serum creatinine over multiple measurements (if available) can be used to judge the degree of decline in GFR.
- If the serum creatinine is rising briskly (or if only a single initial value is available), then the GFR should be presumed to be 0 mL/min, and drugs should be dosed accordingly.
- If the serum creatinine is falling, the eGFR based on the serum creatinine likely underestimates the true GFR. In such cases, the medications should be dosed according to a GFR greater than the calculated eGFR, with re-evaluation of dosing on a daily basis depending upon the trajectory of improvement.
- If the creatinine has reached a plateau and is stable for several days or more, the serum creatinine may be used to estimate GFR.
- Formulas for the GFR only work in steady state (equilibrium) conditions. This usually isn’t the case in acute kidney injury. Instead, the trend in serum creatinine over multiple measurements (if available) can be used to judge the degree of decline in GFR.
6.Avoid giving potassium
- Discontinue potassium supplementations, potassium-sparing diuretics and ACE inhibitors/ARBs in patients with AKI.
- Keep in mind that in contrast to popular dogma, ringer lactate is absolutely safe in hyperkalemia.
7.Correct metabolic effect including “Hyperphosphatemia”
- Restrict dietary phosphorous to <2 g per day in all patients with AKI, except those with hypophosphatemia.
- Initiate phosphate binders in patients with AKI who meet the following requirements:
- PO4 >6 mg/dl.
- Hyperphosphatemic patients with RML or tumor lysis syndrome.
- Hyperphosphatemic patients with AKI who have severe hypocalcemia, even if the phosphate concentration is <6 mg/dl or if RRT initiation is imminent.
- Phosphate binder of choice: The selection of phosphate binder depends on the level of serum ionized calcium concentration as:
- Calcium acetate: If the serum ionized calcium concentration is low, calcium-containing phosphate binders such as calcium acetate may be given to control serum phosphate levels. Calcium acetate can be started as two tablets (667mg tablets) TID with meals.
- Sevelamer: If the serum ionized calcium concentration is high, non-calcium phosphate binders (such as sevelamer) are preferred, in order to prevent increases in the calcium/phosphate product.
- Sevelamer can be started at 800 mg PO TID with meals, double dose if needed.
Dialysis
Indications for emergent dialysis *:
- Intractable electrolyte abnormalities especially refractory hyperkalemia.
- Refractory volume overload causing pulmonary edema with hypoxia.
- Life-threatening uremic symptoms: Patients with AKI who have seizures or severe pericardial effusion, active/major bleeding (e.g. GI bleeding) that are attributed to uremia should undergo urgent RRT.
- Patients who have less severe uremic findings, such as altered mental status, are also frequently treated with RRT unless resolution of AKI is judged to be imminent.
- Drug intoxication (e.g. nephrotoxic drug intoxication amenable to dialysis) such as salicylates, lithium, isopropanol, methanol, and ethylene glycol.
Timing of dialysis in non-emergent AKI
- STARRT-AKI is the largest trial to date comparing immediate/early dialysis with delayed dialysis in AKI patients. It is a multinational RCT of 3019 critically ill patients with AKI (mostly with sepsis), comparing an accelerated strategy (median 6hrs to initiation of dialysis) vs standard strategy dialysis (median 31hrs to initiation of dialysis) with a primary outcome of death from any cause at 90 days.
-
Among critically ill patients with acute kidney injury, starting dialysis early (6hrs) for patients with AKI and no true emergency indications compared to 31hrs (standard strategy), has no mortality benefit *.
AKI in specific conditions
For critically ill patients with AKI, the first priority is resuscitation (explained above). However the ideal treatment is to identify and address the underlying cause of AKI. The above discussion focused on initial resuscitation of AKI in ED. In the following section, AKI in specific conditions will be explored.
Rhabdomyolysis
Rhabdomyolysis (RML) is a syndrome characterized by muscle necrosis and the release of intracellular muscle constituents (e.g. myoglobin, muscle enzymes) into the circulation with the potential to cause AKI. Myoglobin may injure the kidney in three ways *:
- Tubular obstruction, possibly in association with uric acid.
- Direct proximal tubular epithelial cell injury.
- Vasoconstriction, which results in a reduction in blood flow in the outer medulla.
Causes *
A. Traumatic or compression
-Crush injury
-Multiple trauma
-Compartment syndrome
-Coma
-Immobilization
-Electrical injury, burns
-Venoms (snake, spider)
B. Nontraumatic
1. Excessive muscle activity
-Extreme exertion, environmental heat illness.
-Status epilepticus
-Agitation, hyperkinetic states
-Muscle tone disorders e.g. Neuroleptic malignant syndrome, serotonin syndrome.
2. Drugs and toxins
-Statins & fibrates.
-Colchicine
-Antimicrobials: Quinolones, trimethoprim-sulfamethoxazole, amphotericin B, tenofovir, zidovudine
-Toxicology: Alcohol, sympathomimetics
3. Metabolic
-Electrolyte abnormalities: ↓PO4, K, Ca.
-Hyperthyroidism or hypothyroidism
4. Myopathy
-Infections: Viral infections (e.g. influenza, HIV), clostridium spp, Toxic shock syndrome.
-Inflammatory myopathies: Dermatomyositis, polymyositis.
-Metabolic myopathies
Clinical manifestations
- Symptoms: Muscle pain (25% of patients)*, stiffness, swelling, cramping, weakness.
- Signs: Reddish-brown urine, skin changes of ischemic tissue injury such as discoloration or blisters, other signs of muscle damage (e.g. compartment syndrome or ischemic limb).
- Dark urine reflects the presence of toxic myoglobin within the urine, so this is a concerning sign for clinically relevant kidney injury.
Diagnostic evaluation
- Serum CK.
- Muscle enzymes: Aldolase, aminotransferases, LDH.
- Urinalysis, including dipstick and microscopic evaluation.
- Complete blood count and electrolyte, BUN, creatinine, with BUN to Cr ratio (BCR).
- BCR < ~5. Both BUN and creatinine levels are increased in rhabdomyolysis. However there will be a disproportionate rise in creatinine early in the course of the disease due to metabolism of large amounts of CK released by the injured muscles. This causes the BCR ratio to be as low as five or even less *.
- ECG: Evaluate cardiac dysrhythmias related to ↓K and ↓Ca.
Laboratory findings
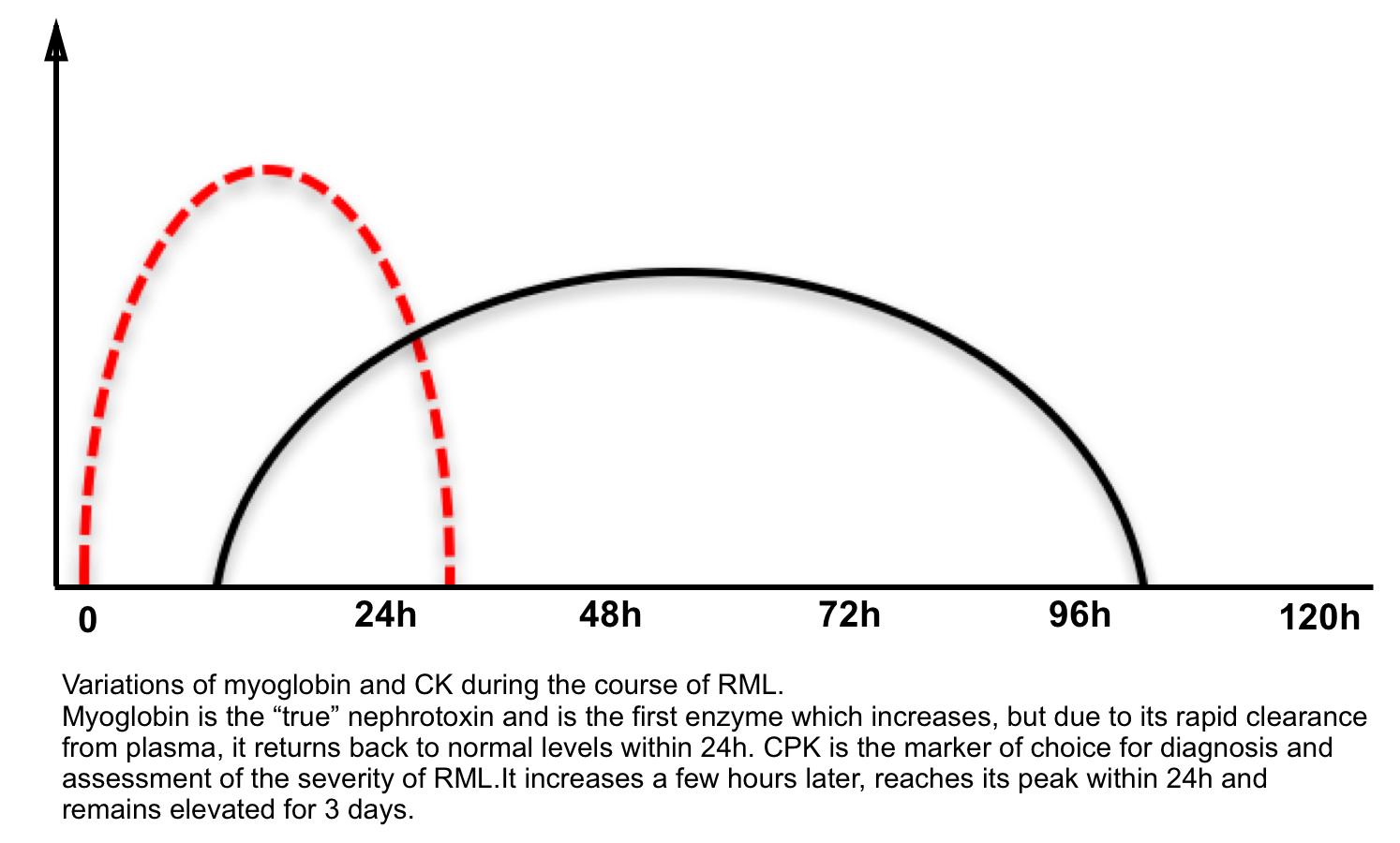
- ↑Serum creatine kinase (CK) is the standard biomarker for RML.
- CK levels at presentation are usually at least 5 times the upper limit of normal (>1000 U/L).
- The serum CK begins to rise within 2-12h after the onset of muscle injury, and reaches its maximum within 24-72h. A decline is usually seen within 3-5 days of cessation of muscle injury.
- Myoglobinuria. Myoglobin is a heme-containing protein.
- It peaks earlier than CK and disappears while CK level is still substantially elevated or rising (due to rapid renal clearance).
- Myoglobinuria lacks sensitivity as a test for rhabdomyolysis; it may be absent in 25-50% of patients with RML due to the more rapid clearance of myoglobin, compared with CK, following muscle injury.
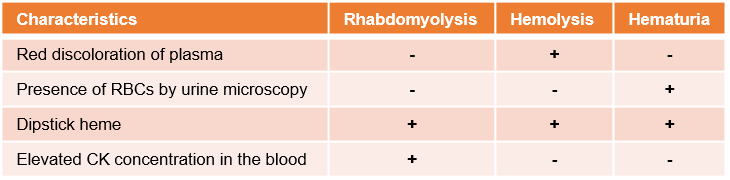
- Both hemoglobin and myoglobin can be detected on urine dipstick as “blood”, however microscopic evaluation of the urine generally shows few-no RBC in patients with RML (table below).
- ↑AST. Elevation of AST alone (or a dramatically ↑AST with minimally elevated ALT) raises a question of AST release from the muscle due to rhabdomyolysis.
- Electrolyte abnormalities. Necrosis of large quantities of tissue may cause the following constellation of electrolyte changes (similar to tumor lysis syndrome):
- ↑K
- ↑PO4
- ↓Ca (may occur in first few days as calcium enters damaged muscle cells, and also forms complexes with phosphate)
- ↑Uric acid and LDH levels.
Diagnosis and differentials
- There is no single consensus definition.
- Diagnosis of RML can be considered in a patient with either an acute neuromuscular illness or dark urine without other symptoms, plus a marked acute elevation in serum CK ( >5000 units/L).
- If RML is highly suspicious but CK level is mild-moderately elevated, serial CK measurement is helpful.
- The differential diagnosis of red urine discoloration is presented below.
Risk of AKI and dialysis
- Serum CK level *
- CK 1000-5000: Low risk of kidney injury.
- CK 5000-15000: Increased risk of kidney injury.
- CK >15000: increased risk of dialysis.
- McMahon score. This is a prognostic score, which estimates severity and requirement for dialysis in rhabdomyolysis patients, which includes age, sex, Cr, Ca, CK, phosphate and bicarb as the score parameters *.
- A score of ≥6 indicates risk of AKI or dialysis, suggesting a possible benefit from treatment.
- Advantages of McMahon score compared to CK level alone:
- This can facilitate treatment without waiting for the CK level to rise above 5,000 U/L.
- The McMahon score is based on more solid evidence than purely CK-based definitions of rhabdomyolysis.
- In one validation study, a McMahon score of 6 or greater had performance superior to CK > 5,000 U/L for the prediction of dialysis (McMahon had sensitivity and specificity of 86% and 68% respectively, whereas CK >5,000 U/L had sensitivity and specificity of only 83% and 55%).
Approach to diagnosis and management
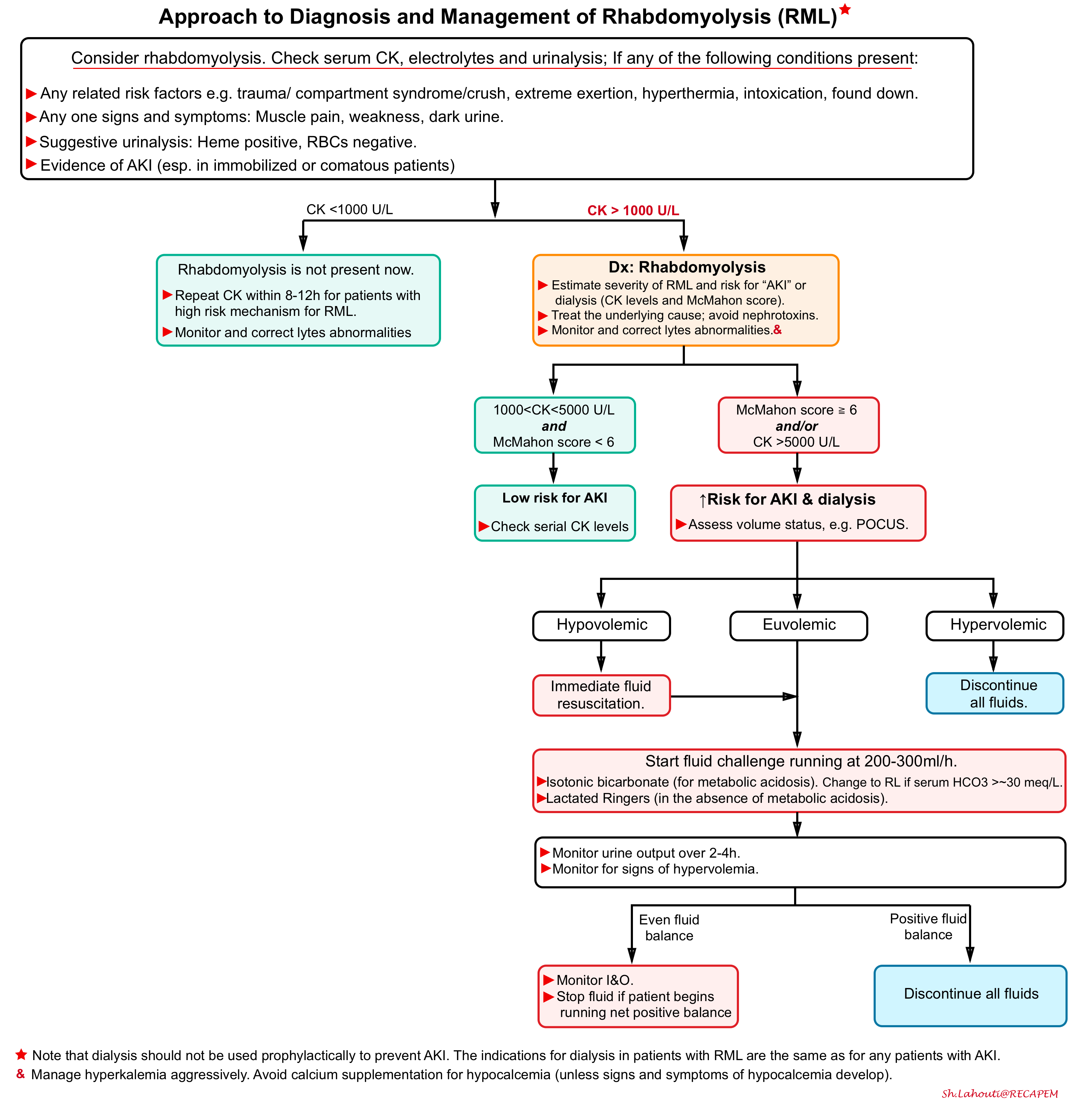
Evaluate and treat underlying causes
- Discontinue myotoxic medications.
- Evaluate for the underlying cause of RML (e.g. ischemic limb, compartment syndrome, status epilepticus).
- Compartment syndrome can cause RML (for example a tibial fracture can cause compartment syndrome and consequently RML establishes if the primary problem is not promptly managed).
- On the other hand compartment syndrome can develop secondary to RML (following marked swelling and edema of the involved muscle groups). Cutaneous signs (e.g. blisters, image below), paresthesia or paralysis should raise the alertness for the possibility of developing compartment syndrome.
Discontinue any nephrotoxic medications
Manage electrolyte disturbances
- Hyperkalemia should be aggressively treated with standard medical management.
- Calcium supplementation for hypocalcemia should be avoided (to minimize the late occurrence of hypercalcemia as well as the risk of calcium phosphate precipitation), unless significant signs and symptoms of hypocalcemia develop or calcium administration is required for the management of hyperkalemia *.
Fluid administration
- Generally, it is believed that aggressive fluid administration will limit intratubular cast formation by diluting the concentration of myoglobin within the tubular fluid, wash out partially obstructing intratubular casts, and increase urinary potassium excretion. However blind administration of large fluid volume can cause hypervolemia, which is harmful.
- Indications for fluid administration:
- Any patients with suspected rhabdomyolysis and clear evidence of hypovolemia should be aggressively rehydrated toward euvolemic state.
- Euvolemic patients requires prophyalctic rehydration only when CK >5000 U/L and/or McMahon score ≥6.
- Volume of fluid
- The optimal rate of repletion is unclear. No studies have directly compared the efficacy and safety of different rates of fluid administration in this setting.
- Following aggressive rehydration in hypovolemic patients, the IV fluid (if indicated) can be given at 4 mL/kg/h with the goal of maintaining a minimum urine output of 3 to 4 mL/kg/h *. Another method is a goal of 200 to 300 mL of urine output each hour *.
- Type of fluid
- No prospective controlled studies have identified ideal fluid choice.
- Lactated Ringers: Reasonable for patients without metabolic acidosis and with relatively normal electrolytes.
- Isotonic bicarbonate: For patients with a non-anion-gap metabolic acidosis or uremic acidosis.
- Goal: increasing the serum bicarbonate to a high-normal level (~ 30 mEq/L).
- Watch for ↓K and ↓Ca.
- Duration of rehydration
- Most guidelines recommend IV rehydration for the next 24-72h, or continuing fluid until the CK is <5,000 U/L. However there’re several caveats here:
- The true nephrotoxin is myoglobin but not CK. However myoglobin is rapidly cleared while CK level remains elevated for days after myoglobin is gone.
- CK has a half-life of 36h. Thus, if the CK is markedly elevated (e.g. 25.000 U/L), it will remain above 5,000 U/L for days, long after myoglobin is gone.
- Hence, the more physiologic approach may involve monitoring patient’s fluid balance rather than strict CK level.
- If the patient is running an even fluid balance (i.e. excreting all the administered fluid), then rehydration isn’t causing harm.
- If the patient is running a positive fluid balance, then consider earlier termination of fluid administration.
- Most guidelines recommend IV rehydration for the next 24-72h, or continuing fluid until the CK is <5,000 U/L. However there’re several caveats here:
Urine alkalization
- In theory, urine alkalization prevents heme protein precipitation with Tamm Horsfall protein and therefore intratubular pigment cast formation. However there is no clear clinical evidence that an alkaline diuresis is more effective than a saline diuresis in preventing AKI.
- Isotonic bicarbonate is reasonable choice in patients with metabolic acidosis.
- Avoid metabolic alkalosis (target serum bicarbonate to ~30 mEq/L).
- Monitor closely for development of hypokalemia and hypocalcemia *.
Avoid hypervolemia
- Fluid overload is nephrotoxic. Stop IV fluid when patient become hypervolemic.
- Diuretic: If there is evidence of volume overload, judicious use of loop diuretics may be justified in patients with RML.
Dialysis: This should not be used to prevent AKI in patients with RML. Indications for dialysis in these patients are the same as for any patients with AKI.
Tumor lysis syndrome
Tumor lysis syndrome is a condition caused by metabolic and electrolyte derangements secondary to rapid cell destruction, usually following chemotherapy for patients with cancer. The pathophysiology and clinical manifestations of TLS is illustrated below.
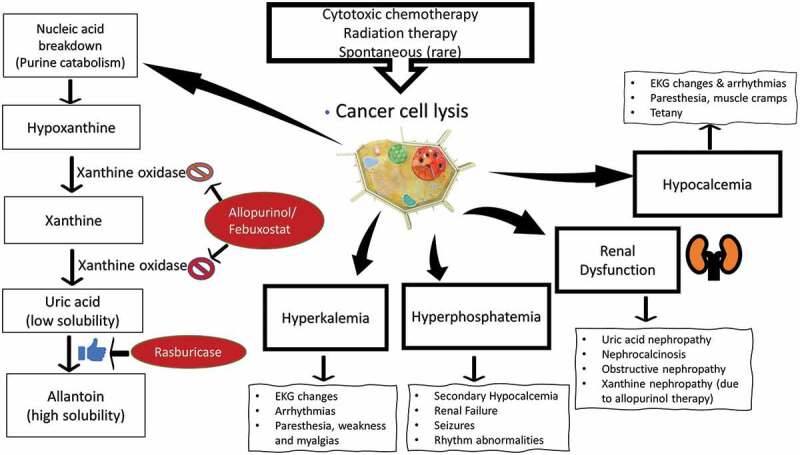
- This syndrome occurs within 1 week after the initiation of the chemotherapy (or irradiation), or it can rarely occur spontaneously.
- Lysis of a large number of cancer cells leads to the release of a significant amount of intracellular contents (DNA, phosphate, potassium, and cytokines) into the systemic circulation.
- Hyperuricemia
- Destruction of tumor cells releases purine nucleic acids. The catabolism of nucleic acids are shown above.
- Uric acid and Xanthine are both potentially nephrotoxic (due to precipitation in the renal tubules); with uric acid having a greater tendency to precipitate and cause renal damage.
- Allopurinol inhibits xanthine oxidase, leading to reduction in uric acid production; however this may cause accumulation of Xanthine (which is potentially nephrotoxic but not as much as uric acid). Patients with established TLS who are receiving allopurinol are at risk for xanthine precipitation in the tubules, resulting in “xanthine nephropathy” or “xanthine stone formation”. Discontinuation of allopurinol is warranted in such patients.
- Rasburicase is the most effective approach to manage hyperuracemia. It is the enzyme that converts uric acid into allantoin, which is harmless. In contrast to allopurinol, treatment with rasburicase has the advantage of not leading to accumulation of Xanthine.
- Only Rasburicase can act on uric acid which has already been generated (allopurinol can prevent uric acid buildup, but doesn’t affect pre-existing uric acid).
- Hyperphosphatemia
- The phosphorus concentration in malignant cells is up to four times higher than in normal cells. Thus, rapid tumor breakdown often leads to hyperphosphatemia, which can cause secondary hypocalcemia, leading to tetany or seizures.
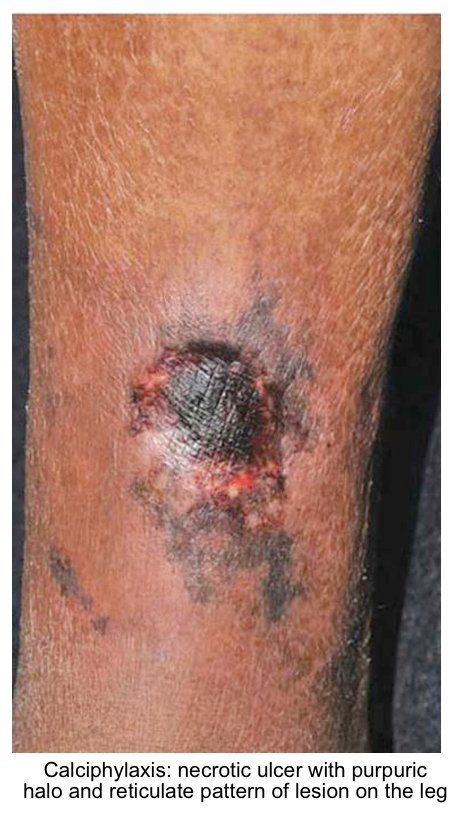
- The calcium-phosphate product is the calcium concentration multiplied by the phosphate concentration (both in mg/dL). When the calcium-phosphate product rises >60 mg2/dL2, there is an increased risk of systemic precipitation of calcium phosphate (calciphylaxis). This may manifest as necrotizing skin ulceration (figure below).
- AKI. The mechanism of AKI in TLS is multifactorial *
- Hyperuricemia can lead to acute renal failure by direct crystallization within the renal tubules. However since the widespread use of hypouricemic agents, calcium phosphate deposition (nephrocalcinosis), rather than hyperuricemia, has become the major mechanism of acute kidney injury in TLS.
- Tumor related high risk features
- Type of the tumor.
- Most common in hematologic malignancies such as leukemia and lymphoma (being most often associated with non-Hodgkin’s lymphoma and acute lymphocytic leukemia).
- May happen in solid tumor with large tumor burden (e.g. neuroblastoma, germ cell cancer, small cell lung cancer).
- Tumors with rapid cellular proliferation rate: LDH is an indicator of rapid cell turnover or tumor lysis, and higher levels equate to higher risk. Baseline LDH more than two times the ULN carries high risk for TLS.
- High tumor burden *:
- Bulky tumor (>10cm )
- Widely metastatic disease
- WBC count >50,000 per microL
- Type of the tumor.
- Treatment related high risk features
- Use of novel and more potent therapies (e.g. biological treatments).
- Inadequate hydration during cytoreductive therapy.
- Use of nephrotoxins.
- Patient related risk factors:
- Pre-existing renal disease /uremia.
- Pretreatment hyperuricemia (uric acid >7.5 mg/dL).
- Pretreatment hyperphosphatemia (serum phosphate >4.5 mg/dL).
Clinical manifestations. Signs and symptoms of TLS are usually the result of electrolyte derangement
- ↑K 👉Arrhythmias, sudden death.
- ↓Ca👉 Paresthesia, muscle cramps/tetany, seizures, delirium, hypotension, cardiac arrhythmia.
- ↑PO4 👉Nausea, vomiting, diarrhea, lethargy, and seizures.
- Hyperuricemia👉Oliguric renal failure.
- Urinalysis may classically shows many uric acid crystals or amorphous urates in an acid urine.
Diagnosis: The modern definition of TLS is based on the Cairo–Bishop criteria for laboratory and clinical TLS *.
- Laboratory diagnosis: Malignancy plus ≥two of the following abnormalities (1)
- K >6 mEq/L.
- PO4 >4.5 mg/dL in adults (or >6.5 mg/dL in children).
- Uric acid >8 mg/dL.
- Corrected calcium <7 mg/dL, (or ionized calcium<1.2).
- Clinical definition: Laboratory definition plus at least one of the following:
- AKI (Cr >1.5 x baseline).
- Cardiac arrhythmia.
- Sudden death.
- Seizure.
(1)The definition may also be met based on changes in electrolytes over time (e.g. 25% increase in potassium, 25% increase in phosphate, or 25% decrease in calcium).
Differential diagnosis
- Rhabdomyolysis
- Hemolysis
- Fulminant hepatitis
- Pseudohyperkalemia (may occur among patients with very high WBC or PLT count).
Prevention. Begins with identifying patients who are at risk for TLS prior to treatment.
- Monitoring
- Patients should be monitored before and during their first 7d of anti-cancer therapy with labs (uric acid, BUN, creatinine, calcium, phosphate, and LDH).
- Hight risk patients should be monitored 4-6h after the initiation of chemotherapy and every 4-8h thereafter *.
- Renal protection
- Avoid nephrotoxic medications
- Avoid renal malperfusion (avoid hypotension, and volume overload).
- Hydration
- Goal: Establishing high urine output while maintaining euvolemia (e.g. urinary output >100 mL/h for the 24 h before chemotherapy initiation and throughout treatment duration).
- Choice of fluid:
- Generally balanced crystalloid (e.g. RL, plasmalyte) is preferred.
- For patients with uremic acidosis or NAGMA, isotonic bicarbonate is reasonable.
- Hypouracemic agents. Choice of the regimen depends on risk stratification for TLS.
- High risk for TLS
- Rasburicase prophylaxis is recommended. Dosing: 0.2 mg/kg once daily for up to 5-7 days.
- Intermediate risk
- Allopurinol (as long as uric acid is not elevated i.e. <8 mg/dL). It may be started 2-3 days before chemotherapy. The usual dose of allopurinol is 300 mg PO BID. The dose must be reduced by 50% in the setting of AKI.
- Febuxostat. It is another xanthine oxidase inhibitor that may be used for patients allergic to allopurinol. Dosing: 40 to 80 mg once daily or 120 mg once daily.
- Low risk: hydration alone is recommended.
- High risk for TLS
Treatment of established TLS
- Admit the patient to the ICU with continuous cardiac monitoring and frequent laboratory measurements.
- Treat hyperkalemia promptly.
- Limit intake of potassium and phosphate intake.
- Avoid calcium administration for asymptomatic hypocalcemia.
- Additional calcium may continue to precipitate with phosphorus, accumulating in the renal tubules and further worsening renal injury.
- Only treat hypocalcemia-induced seizures, tetany, and ECG changes.
- Simply controlling phosphate levels can prevent hypocalcemia.
- Provide phosphate binder for hyperphosphatemia.
- Give Sevelamer 800-1600 mg TID with meals. If phosphate level is >9 mg/dL, use a higher dose.
- Avoid nephrotoxins.
- Defend renal perfusion.
- Avoid urinary alkalization.
- This approach has fallen out of favor. Although urine alkalization increases the solubility of uric acid, it also decreases the solubility of phosphate and may precipitate calcium phosphate in the renal tubules, further worsening the AKI *.
- Hyperuracemia 👉Rasburicase
- Indicated for patients with TLS and a uric acid level of >8 mg/dL (especially patients with acute renal failure).
- It reduces uric acid levels within 4 h of administration *. The half-life of rasburicase is ~20 h, therefore giving a single dose per day is recommended.
- Contraindications: G6PD deficiency, and pregnant patients. See Black Box Warnings.
- Dosing: 0.2 mg/kg/day IV over 30 min q24h for up to 5 d. However, most hospitals have a single-dose protocol with doses ranging from 3-9 mg per day.
- Discontinue allopurinol (see above). Allopurinol is not only ineffective in the treatment of existing hyperuracemia, rather it is harmful by increasing production of Xanthine and related risk of Xanthine nephropathy. Allopurinol may also reduce the efficacy of rasburicase, so the two drugs should not be used together.
- Hydration. Goal is to establish high urine output while maintaining euvolemia.
- If patient is hypovolemic, give balanced crystalloid e.g. RL to achieve euvolemia.
- For patients with uremic acidosis or NAGMA, isotonic bicarbonate is preferred. Keep serum bicarb at low-normal range e.g. 20-24 mmol/L.
- Once patient is euvolemic, provide adequate amount of IV fluid to keep urinary output of at least 100-200ml/h *.
- If patient is not excreting most of the given fluid, consider judicious administration of diuretic. Furosemide is preferred over thiazide diuretic (which tend to increase serum uric acid).
- If the patient has poor urine output despite diuretic use, then stop fluid administration.
- If patient is hypovolemic, give balanced crystalloid e.g. RL to achieve euvolemia.
- Dialysis. Indications are similar to those in patients with other causes of AKI, although somewhat lower thresholds are used for patients with TLS because of potentially rapid “K” release and accumulation, particularly if urine output is low. Indication for RRT may include *, *:
- Volume overload.
- Uric acid severely elevated despite rasburicase (e.g. >10 mg/dL).
- Marked hyperphosphatemia, elevated Calcium-Phosphate product (e.g. >70 mg2/dL2), symptomatic hypocalcemia.
- Uncontrolled hyperkalemia.
- Uremia, acidosis.
- TLS plus oliguria/anuria.
- Intractable fluid overload.
Abdominal compartment syndrome
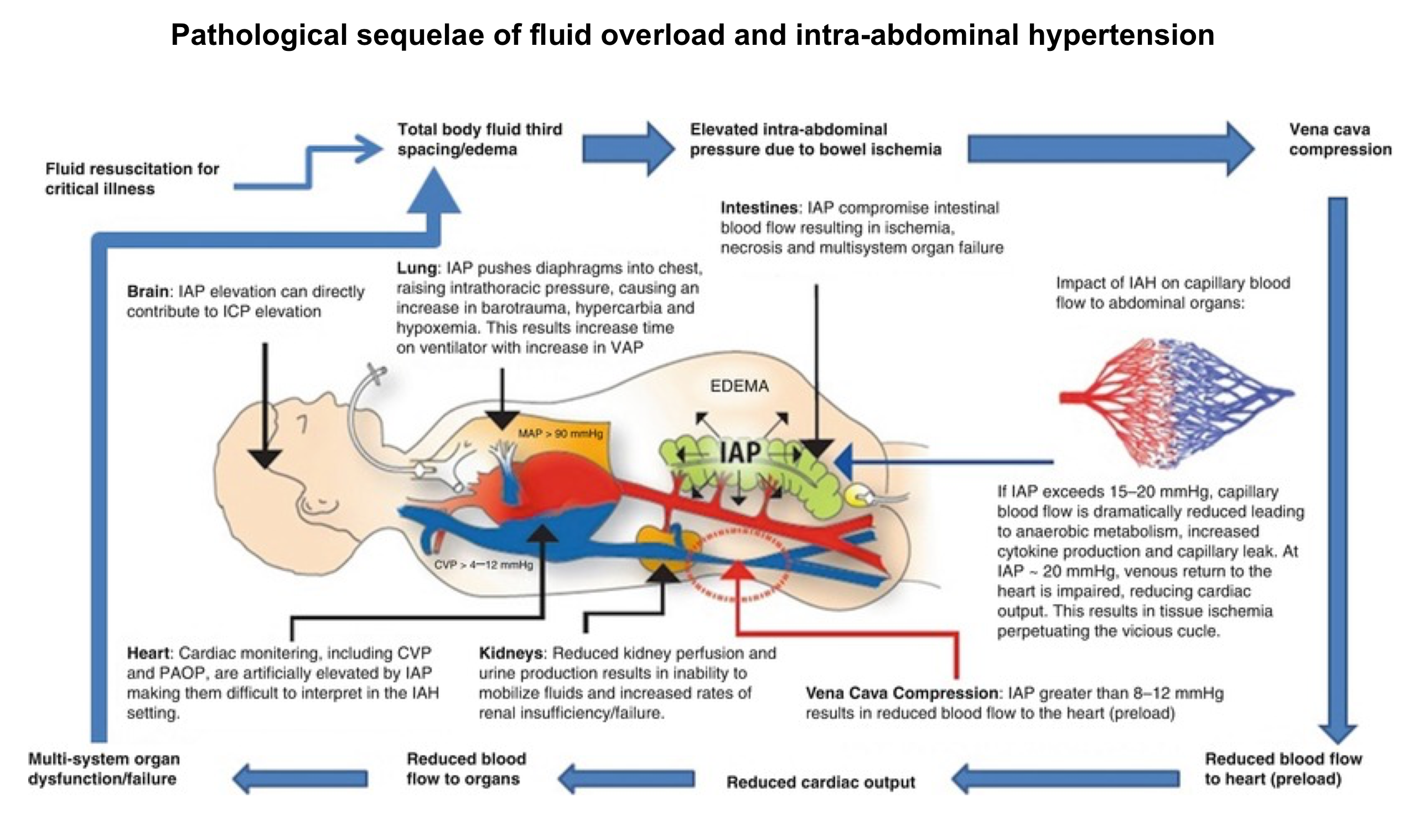
- The effects of intra-abdominal hypertension are not limited just to the intra-abdominal organs, but rather have an impact either directly or indirectly on every organ system in the body (shown below).
- Renal effect: Kidney is most sensitive organ to raised intra-abdominal pressure.
- Direct renal parenchymal compression diminishes renal arterial flow while the renal venous pressure and renal vascular resistance are elevated (aka. “renal compartment syndrome”). This causes blood to be shunted away from the renal cortex and glomeruli leading to impaired glomerular and tubular function in effect triggering renal ischemia and subsequent renal failure (oliguric renal failure).
- Renal effect: Kidney is most sensitive organ to raised intra-abdominal pressure.
Risk factors
- Elevated intra-thoracic pressure (mechanical ventilation, ARDS on high PEEP)
- Morbid obesity
- Fluid resuscitation
Causes
- Intra-abdominal (primary) causes: Ascites, severe pancreatitis, toxic megacolon, ileus, abdominal surgery/trauma.
- Extra-abdominal (secondary) causes: Large volume fluid resuscitation, septic shock, major burn injuries.
Diagnosis
- Abdominal palpation is highly insensitive for detection of abdominal compartment syndrome, however presence of tense abdomen is relatively specific (~80%) for abdominal compartment syndrome.
- Intra-vesicular pressure measurement is simple, precise, and minimally invasive method for measurement of intra-abdominal pressure.
- In an average adult, normal intra-abdominal pressure (IAP) is within the range of 0–5 mmHg.
- IAP ≥12 mmHg, is used to define intra-abdominal hypertension
- IAP > 20 mmHg is the cutoff used to describe abdominal compartment syndrome.
- Diagnostic criteria of abdominal compartment syndrome:
- Sustained IAP pressure >20 mm Hg.
- New organ dysfunction/failure attributable to elevated intra-abdominal pressure.
Treatment
- Defend abdominal perfusion pressure (APP). {APP = MAP-IAP}
- By definition, APP is the difference between mean arterial pressure (MAP) and the IAP. This is the pressure that drives perfusion of all intra-abdominal organs (e.g. the kidney).
- An APP< 60 mmHg has been shown to reliably predict the necessity for surgical decompression, making it useful both in diagnosis and treatment.
- Target MAP to > (60 + intra-abdominal pressure). Employ vasopressors to achieve the target MAP, and avoid additional fluid *.
⚠️Caution: Ultrasound evaluation of IVC is not reliable in the presence of intra-abdominal hypertension especially with tense ascites is present. Abdominal compartment syndrome may compress the IVC, making it look empty (image below). This may lead to incorrect decisions regarding fluid administration. Avoid fluid administration since it worsens the interstitial edema and organ perfusion.
- Decompress the abdomen
- Ascites: therapeutic paracentesis.
- NPO, Gastric tube to suction.
- Decompress the colon (e.g. suppositories, neostigmine for megacolon).
- Fascial release is definitive treatment, but most invasive. Reserve this for failure of other measures.
- Decompress the thorax
- Large pleural effusion: Therapeutic thoracentesis.
- Avoid intubation if possible
- Reduce airway pressures as able (e.g. target low PEEP and plateau pressures).
Hepatorenal syndrome
The hepatorenal syndrome (HRS) is one of many potential causes of AKI (see below) in patients with acute or chronic liver disease. Affected patients usually have portal hypertension due to cirrhosis, severe alcoholic hepatitis, but can also have fulminant hepatic failure from any cause.
HRS is possibly the etiology of 10-30% of cases of acute kidney injury among patients with cirrhosis *. It occurs in advanced liver disease patients *
- 20% of patients within 1 y.
- 40% of patients within 5 y.
Definition
- Hepatorenal Syndrome (HRS) is defined as a deterioration of kidney function that takes place in the context of severe chronic liver diseases, such as advanced cirrhosis or acute liver failure.
- HRS is divided into two types:
- Hepatorenal Syndrome Type-1 (recently renamed as HRS-AKI *): Acute kidney injury due to hepatorenal physiology (will be discussed here).
- Hepatorenal Syndrome Type-2 ( recently renamed as HRS-CKD *): Gradual kidney function impairment (due to hepatorenal physiology) that is less severe than that observed with type 1 disease.
- The major clinical feature in patients with type 2 hepatorenal syndrome is ascites that is resistant to diuretics.
Pathophysiology of HRS-1
- Liver failure causes systemic vasodilation due to a release of vasodilators (e.g. nitric oxide, prostacyclins) from hepatic circulation *.
- This causes marked reduction of effective arterial blood volume resulting from blood volume redistribution into the splanchnic circulation and relatively insufficient cardiac output (and low blood pressure). This leads to renal hypoperfusion caused by reduced renal perfusion pressure.
- The compensatory response to this systemic vasodilation, is activation of the sympathetic nervous system and the renin-angiotensin aldosterone system (releasing endogenous norepinephrine and angiotensin II).
- Initially, these systems are able to compensate. But beyond a certain point, patients develop refractory systemic vasodilation. Unfortunately, endogenous norepinephrine and angiotensin-II remain successful at causing renal vasoconstriction. This will eventually result in systemic vasodilation with renal vasoconstriction, causing inadequate renal perfusion.

- If HRS is recognized early and associated hemodynamic abnormalities are treated, the HRS is expected to resolve. As such, it may be conceptualized as a reversible form of pre-renal renal failure.
- However, if HRS is left untreated, hypoperfusion may eventually lead to dysfunction of the renal tubules (acute tubular necrosis), with intrinsic renal failure.
Risk factor for development of HRS-AKI *
- Advanced cirrhosis with portal hypertension (or acute liver failure)
- Deterioration in liver function (e.g. acute-on-chronic liver failure)
- The greatest risk is among patients with chronically borderline perfusion. Clinically, this may be revealed by:
- Chronic hypotension.
- Chronic hyponatremia *.
- More severe ascites (e.g. refractory ascites) *
Precipitating factors. Well-known causes of circulatory dysfunction and subsequent renal hypoperfusion include
- Infections e.g. spontaneous bacterial peritonitis
- Large volume paracentesis without plasma expansion
- GI bleeding
Intravascular volume depletion (i.e. diuretic-induced, extrarenal fluid losses) has classically been considered as a triggering factor for HRS. However, no convincing evidence has yet been reported to support this pathogenetic relation.
- Although overly rapid diuresis has often been mentioned as a precipitant of hepatorenal syndrome, perhaps because most patients are taking diuretics when the syndrome is diagnosed, diuretics do not cause hepatorenal syndrome.
- Diuretics can, however, cause azotemia, particularly if fluid is removed too rapidly in patients without peripheral edema.
- Diuretic-induced azotemia improves with the cessation of therapy and fluid repletion.
- In comparison, the HRS typically worsens progressively, even after diuretics are stopped and albumin is infused.
Prevention of HRS
- Avoid nephrotoxins in patients with advanced cirrhosis (esp. NSAIDs, ACE inhibitors, and ARBs); if possible.
- Albumin. In the following conditions, albumin administration has been shown to reduce the risk of HRS.
- Large volume paracentesis (LVP). Administration of 8g albumin per liter of fluid removed will reduce the risk of HRS.
- Spontaneous bacterial peritonitis (SBP). It has been shown that albumin administration dramatically reduce risk of HRS and mortality *.
- The treatment regimen is 1.5 g/kg of 20% albumin at the time of diagnosis of infection, and another dose of albumin (1 g/kg) 48h later.
- Antibiotic. A randomized trial reported significant benefits with the oral administration of norfloxacin at 400 mg/day to patients with cirrhosis and ascitic fluid total protein <1.5 g/dL who fulfilled either of the following two criteria: a Child-Pugh score >9 points and serum bilirubin >3 mg/dL; or a serum creatinine >1.2 mg/dL or BUN >20 mg/dL or serum sodium <130 mEq/L *. Alternative antibiotic include, trimethoprim-sulfamethoxazole (one double-strength tablet daily), ciprofloxacin (500mg per day).
Clinical presentation
In a patients with established or clinically evident acute or chronic liver disease; HRS presents in a similar fashion to any other form of renal failure due to malperfusion, typically:
- A progressive rise in serum creatinine. Interpretation of serum creatinine in patients with cirrhosis is tricky! Patients with HRS may have kidney dysfunction that is substantially more severe than is suggested by the serum creatinine.
- Both urea and creatinine production may be substantially reduced in the setting of liver disease or due to decreased muscle mass and decreased protein and meat intake. The net effect is that a serum creatinine that appears to be within the normal range (e.g. 1 to 1.3 mg/dl) may be associated with a GFR that ranges from as low as 20 to 60 mL/min. Therefore creatinine level tends to overestimate the patient’s renal function (see above).
- Small changes in creatinine may be significant. Serum creatinine may increase by as little as 0.1 mg/dl per day, with intermittent periods of stabilization or even slight improvement. For example, if the creatinine rises from a baseline of 0.5 mg/dl to 0.8 mg/dl, this may reflect a major reduction in renal function.
- Oliguria (e.g. daily urine output < 400-500 ml).
- However, many patients with HRS are nonoliguric (especially early in the course). Remember that oliguria may be a late finding in renal insufficiency. Some studies have found that the urine volume may exceed 400 mL per day, with markedly lower output being observed only within a few days from death *.
- Other signs and symptoms related to decompensated chronic liver disease or acute liver failure such as:
- Borderline/low BP. Cirrhosis or acute liver failure causes systemic vasodilation resulting in low BP. In fact MAP > 80 mmHg argues against HRS.
- Hypoglycemia, due to inability of the liver to synthetize glucose.
- Dyspnea can be present due to massive ascites and/or hydrothorax.
- Delirium can be caused by hepatic encephalopathy.
- GI bleeding secondary to variceal hemorrhage may be present.
Differential diagnosis of AKI in patients with liver disease
- Prerenal azotemia
- Hypovolemia (GI hemorrhage, GI losses from lactulose induced diarrhea, over-diuresis).
- Shock from any etiology.
- Hypervolemia (congestive hepatopathy).
- Tense ascites with abdominal compartment syndrome.
- Intrinsic AKI
- Ischemic ATN (severe hypoperfusion).
- Nephrotoxic medications.
- Glomerulonephritis or vasculitis e.g. vasculitis accompanied with chronic HBV or HCV infection.
Evaluation
- Review of medications
- Urinalysis
- Urine protein and electrolytes.
- Note: Urinary sodium and FeNa are no longer part of the diagnostic criteria of AKI-HRS.
- Assessment of bacterial infection e.g. paracentesis, blood and urine culture etc.
- Leukocytosis may be absent owing to hypersplenism in patients with cirrhosis and infection *.
- Renal ultrasound *
- Renal ultrasonography is the modality of choice to rule out anatomical abnormalities, such as hydronephrosis, that would negate a diagnosis of HRS-1 as the primary cause of AKI.
- Hemodynamic evaluation. In patients with cirrhosis, ultrasound findings can be confounded and ambiguous. In the presence of equivocal ultrasonographic findings, it’s better to rely on clinical and historical clues. Make your clinical decision on US findings only when these are unequivocal.
- For example while a small and collapsible IVC may suggest hypovolemia with potential implication for volume resuscitation; a collapsible IVC in the presence of tense ascites is suggestive for intra-abdominal hypertension (see the figure above). Such patients benefit from therapeutic paracentesis to reduce their intra-abdominal pressure and thereby improving renal perfusion.
- Some patients with cirrhosis develop systemic congestion with elevated right atrial pressure (congestive nephropathy). A dilated IVC plus other related findings of congestion on Doppler ultrasound of abdominal veins support the diagnosis. Such patient may benefit from a diuretic trial.
Diagnosis
The diagnosis of the HRS is one of exclusion, entertained only after other potential causes of acute or subacute kidney injury have been ruled out *. The current diagnostic criteria requires *,*
-
Chronic or acute hepatic disease with advanced hepatic failure and portal hypertension.
- Acute kidney injury, defined as (1)
- An increase in creatinine of >0.3 mg/dl within 48 hours; OR
- An increase from baseline of ≧50% within seven days *.
- The absence of any other apparent cause(s) for the AKI, including
- Absence of shock.
- No recent use of nephrotoxic drugs.
- No response to diuretic withdrawal and a two-day fluid challenge with 1 gram/kg/day of 20-25% albumin to a maximum of 100 grams/day.
- No signs of structural kidney injury *
- Bland urinary sediment.
- Absence of proteinuria (<500 mg/day).
- Absence of microhematuria (>50 RBCs per high-power field); when no urinary catheter is in place.
- Absence of ultrasonographic evidence of obstruction or parenchymal kidney disease.
- No improvement in renal function following discontinuation of of potential nephrotoxins, cessation of antihypertensive medications (if prescribed). Some potential nephrotoxins in this context may include (see the list here):
- Anti-hypertensive medications. Many patients with cirrhosis have hypertension and are treated with antihypertensive medications. Although “cirrhosis cures hypertension” , antihypertensive therapy is continued in many patients and, in conjunction with systemic vasodilation due to cirrhosis, contributes to low MAP.
- Beta blockers are also commonly used in patients with cirrhosis to prevent variceal hemorrhage. However, these agents may be hazardous in patients with ascites and hypotension *, and their discontinuation may lead to resolution of apparent HRS.
(1)The definition of AKI is based on KIDGO criteria (stage 1). Once AKI is diagnosed further staging can be performed as:
- Stage 1A: Serum creatinine <1.5mg/dl.
- Stage 1B: Serum creatinine > 1.5mg/dl.
Pitfall of current approach to HRS
A key element of the traditional approach to diagnosis of HRS is to first provide the patient with a volume challenge of albumin for two days and monitoring the response. If the patient responds to volume loading, then they are diagnosed with pre-renal kidney failure, and if the patient fails to respond to volume loading, then they are diagnosed with HRS. Arguably, this approach is problematic for several reasons:
- Treatment of HRS is more likely to be successful if initiated earlier, when the creatinine is lower. Delaying treatment for two days may impair outcomes.
- Several mechanisms can cause “AKI in cirrhosis” with considerable overlap (figure below). HRS may often co-exist with other renal insults. Therefore, a binary, dichotomous attempt to parse these out as a distinct conditions is erroneous especially among more complex patients.

Diagnostic and therapeutic approach to HRS
The following algorithm represents a possible accelerated approach to hepatorenal syndrome. This will facilitate the clinical reasoning and decision-making for patients with cirrhosis and AKI *. The principles of the following approach involve:
- Albumin administration is based on hemodynamic evaluation.
- Rather than giving a fixed dose of albumin to every patient, volume status is determined clinically (history, exams and POCUS). This will allow a more clinically based approach. For example some patients with a marked hypovolemia may require more aggressive volume resuscitation, while some other patients with hypervolemia (congestive nephropathy) may benefit from diuresis.
- Vasopressor can be started earlier.
- For patients with “possible” HRS, vasoconstrictor therapy can be started earlier (well before the traditional delay of >48 h). Earlier initiation of treatment is more likely to be successful *.
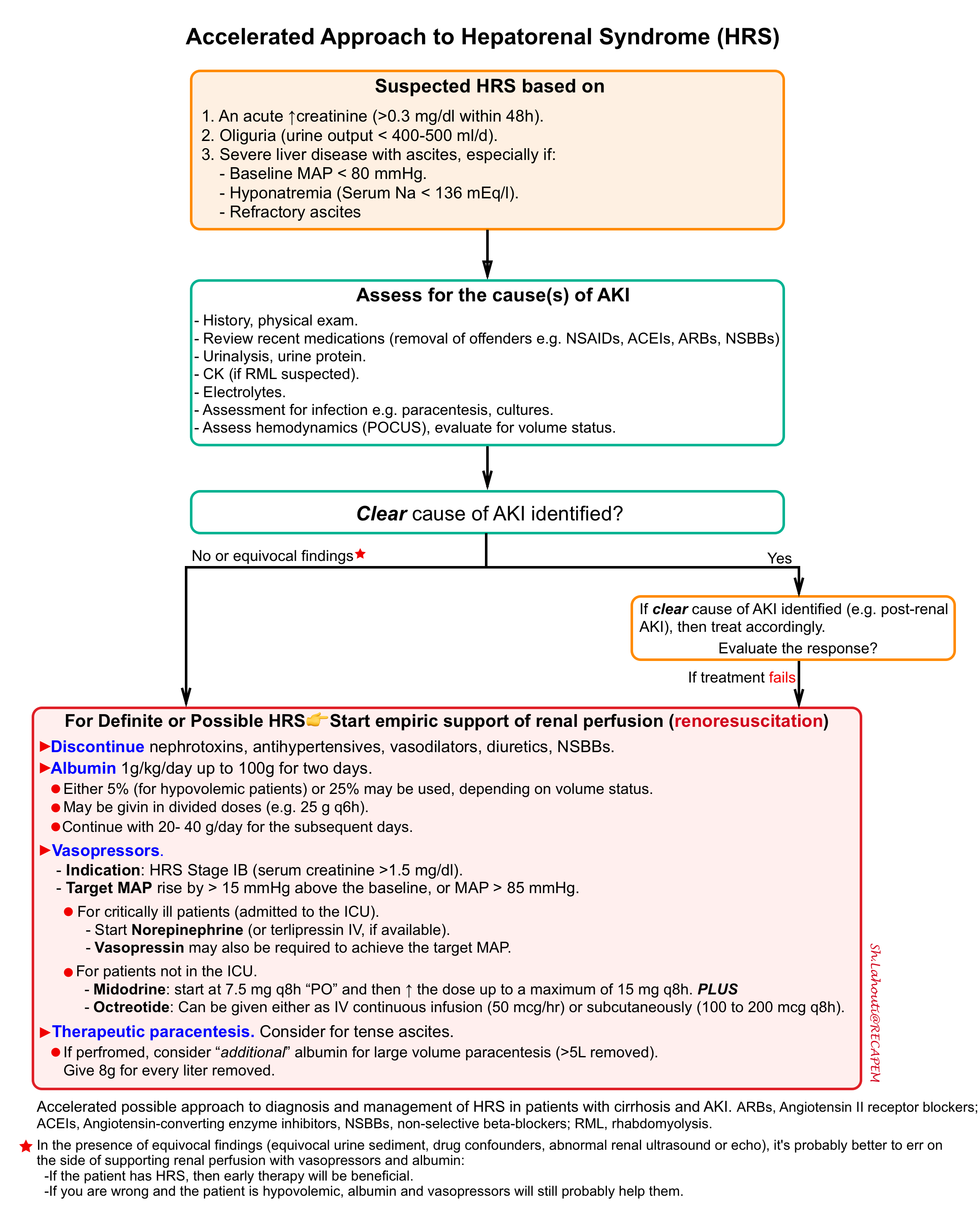
Approach to therapy.
- The ideal therapy for HRS is improvement of liver function from recovery of alcoholic hepatitis, treatment of decompensated hepatitis B with effective antiviral therapy, recovery from acute hepatic failure, or liver transplantation.
- Patients with cirrhosis, ascites, and type I or type II HRS should have an expedited referral for liver transplantation *.
- However, when improvement of liver function is not possible in the short term, the medical therapy should be instituted in an attempt to reverse the AKI associated with HRS.
Vasopressors.
- Rationale.
- The hypovolemia of type 1 HRS is an effective not an absolute volume deficiency. A critical amount of the circulating volume becomes pooled in the enlarged splanchnic circulation.
- The objective of vasoconstriction is to counterbalance splanchnic arterial vasodilation to redistribute part of the splanchnic volume back to the systemic circulation in order to improve renal perfusion *.
- Several meta-analyses have suggested a beneficial effect of the combination of a vasoconstrictor and albumin on short-term survival and HRS *,*,*.
- Indication: Initiate vasopressors for patients with HRS Stage IB (serum creatinine >1.5 g/dL) since more mild forms are likely to resolve with fluid expansion alone *.
- Hemodynamic goal: A target might be a rise of MAP >15 mmHg above the baseline or MAP > 85 mmHg *.
- Agents:
- Terlipressin.
- A meta-analysis that included nine randomized studies (394 patients) comparing different vasoconstrictors in combination with albumin concluded that terlipressin was the most effective in treating HRS *.
- It is administered IV bolus injections; at an initial dose of 0.5 mg to 1 mg q4-6h. The dose can then be gradually increased to a maximum of 2 mg q4-6h if the serum creatinine level does not fall by more than 25% *.
- Norepinephrine:
- It has been found as effective as terlipressin (It is the pressor of choice for patients admitted to the ICU). Indeed, no difference in the reversibility or relapse of HRS was found between the terlipressin + albumin and norepinephrine + albumin arms *.
- Norepinephrine can be given as continuous IV infusion and has the advantage of rapid titration over a broad dose range, in order to promptly achieve BP targets.
- Usual dose range: 0.5 to 3mg/h (8 to 50 mcg/min) *.
- Vasopressin.
- May also be effective in HRS *. but has not been investigated in this context.
- Studies in septic shock have suggested that vasopressin may have a slight advantage over norepinephrine in the preservation of renal function during vasodilatory shock states* .
- It can be started at 0.01 units/min IV and titrating upward (up to 06 U/min ) as needed to raise the MAP *. It should be given through central line.
- {Midodrine plus octreotide}
- The combination of midodrine and octreotide is less effective than terlipressin or norepinephrine *.
- Midodrine forms an active metabolite, that is an alpha1-agonist, and exerts its actions via activation of the alpha-adrenergic receptors of the arteriolar and venous vasculature *.
- Midodrine ca be started at 7.5 mg PO, and increasing the dose at eight-hour intervals up to a maximum of 15 mg PO three times daily) *.
- Octreotide induces a potent splanchnic vasoconstriction probably mediated in part by an inhibition of glucagon release *.
- Octreotide is either given as a 50mcg/h continuous IV infusion or as 100-200 mcg subcutaneously three times daily *.
- Terlipressin.
- Duration of treatment
- The optimal duration of IV vasopressor therapy is unknown, with clinical studies varying between 5-14 days. Vasopressors should perhaps be continued for a longer durations (up to one month or more) until the renal function has improved and stabilized (e.g. returned to baseline or reached a plateau value).
- In patients who respond to therapy, if there is difficulty weaning off vasopressors; then transitioning from IV vasopressor infusion to oral midodrine may be considered.
- Many patients who recover from type 1 HRS continue to have hypotension and refractory ascites, and midodrine may be effective in such patients. By contrast, if a patient has no improvement in kidney function after two weeks, therapy with these drugs can be considered futile.
Albumin
- Rationale. Combining albumin with vasopressor is recommended as front-line therapy for HRS. The beneficial effect of albumin may be explained by * ,*
- Improving hemodynamic and cardiac output via volume expansion.
- Antioxidant effect via possibly adsorbing some circulating toxin including reactive oxygen species, nitric oxide.
- Anti-inflammatory effect.
- Promoting the function and preserving the integrity of endothelial glycocalyx *.
- Dose: 1g/kg/d (up to 100g/d) for two days. It can be also given in divided doses (e.g. 25g q6hr).
- Subsequently, the dose may be decreased to 20-40 grams per day.
- Studies have generally utilized 5% albumin. However, for patients who appear euvolemic or hypervolemic, 25% albumin is probably better.
Therapeutic paracentesis
- Among patients with tense ascites, therapeutic paracentesis is reasonable, if there is concern for intra-abdominal hypertension.
- Additional albumin should be given, if large volume paracentesis(>5L removed) is performed. Give 8g albumin for every liter removed.
Hemodialysis
- Patients with HRS who progress to kidney failure can be treated with RRT, which is most commonly done when patients are awaiting a liver transplant or when there is the possibility of improvement in liver function.
- Patients with an acute and potentially reversible hepatic insult may particularly benefit from dialysis since kidney function will recover in parallel with improving hepatic function.
- Hemodialysis is frequently difficult to perform in patients with HRS since decompensated hepatic function is associated with hypotension and endothelial dysfunction (causing difficulty retaining fluid within their vasculature). Hemodialysis in such cases can precipitate cardiac arrest and death, thereby shortening rather than prolonging survival. Some success has been realized with continuous renal replacement therapy (CRRT).
Media-1
Going further
- Emergency medicine cases podcast. AKI Part 1 and Part 2.
- AKI & oliguria (IBCC)
Appendix
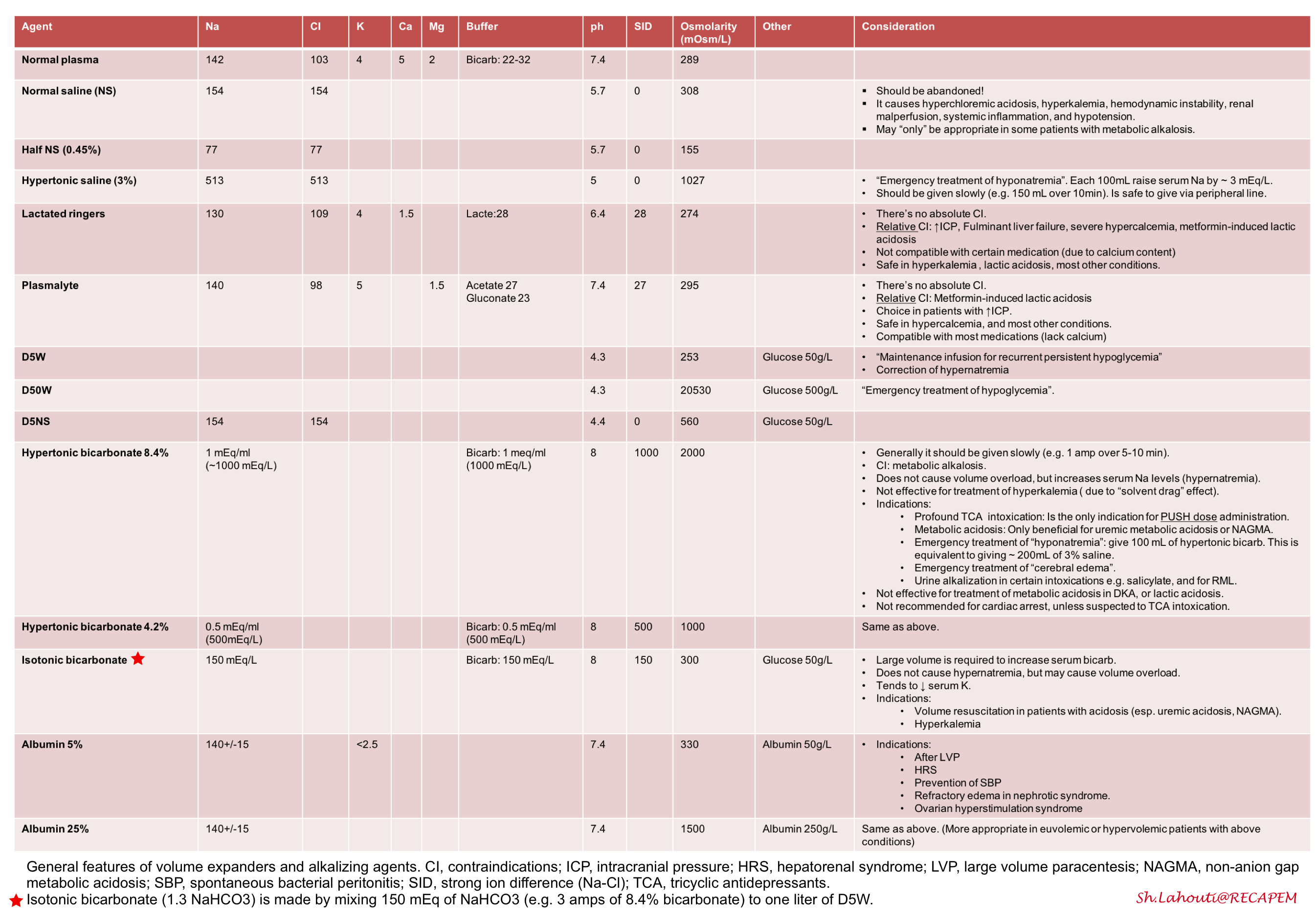
References
PMID: 22890468. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120(4):c179-84. doi: 10.1159/000339789. Epub 2012 Aug 7.
PMID: 25568178. Kellum JA, Sileanu FE, Murugan R, Lucko N, Shaw AD, Clermont G. Classifying AKI by Urine Output versus Serum Creatinine Level. J Am Soc Nephrol. 2015 Sep;26(9):2231-8. doi: 10.1681/ASN.2014070724. Epub 2015 Jan 7.
PMID: 27670788. Ostermann M, Joannidis M. Acute kidney injury 2016: diagnosis and diagnostic workup. Crit Care. 2016 Sep 27;20(1):299. doi: 10.1186/s13054-016-1478-z.
PMID: 19389851. Doi K, Yuen PS, Eisner C, Hu X, Leelahavanichkul A, Schnermann J, Star RA. Reduced production of creatinine limits its use as marker of kidney injury in sepsis. J Am Soc Nephrol. 2009 Jun;20(6):1217-21. doi: 10.1681/ASN.2008060617. Epub 2009 Apr 23.
PMID: 25317932. Thomas ME, Blaine C, Dawnay A, Devonald MA, Ftouh S, Laing C, Latchem S, Lewington A, Milford DV, Ostermann M. The definition of acute kidney injury and its use in practice. Kidney Int. 2015 Jan;87(1):62-73. doi: 10.1038/ki.2014.328. Epub 2014 Oct 15.
PMID: 27670788. Ostermann M, Joannidis M. Acute kidney injury 2016: diagnosis and diagnostic workup. Crit Care. 2016 Sep 27;20(1):299. doi: 10.1186/s13054-016-1478-z.
PMID: 26336912. Lin J, Fernandez H, Shashaty MG, Negoianu D, Testani JM, Berns JS, Parikh CR, Wilson FP. False-Positive Rate of AKI Using Consensus Creatinine-Based Criteria. Clin J Am Soc Nephrol. 2015 Oct 7;10(10):1723-31. doi: 10.2215/CJN.02430315. Epub 2015 Sep 3.
PMID: 9486997.Klahr S, Miller SB. Acute oliguria. N Engl J Med. 1998 Mar 5;338(10):671-5. doi: 10.1056/NEJM199803053381007.
PMID: 21918558. Cerda J. Oliguria: an earlier and accurate biomarker of acute kidney injury? Kidney Int. 2011 Oct;80(7):699-701. doi: 10.1038/ki.2011.177.
PMID: 21918558.Cerda J. Oliguria: an earlier and accurate biomarker of acute kidney injury? Kidney Int. 2011 Oct;80(7):699-701. doi: 10.1038/ki.2011.177.
PMID: 21716258. Macedo E, Malhotra R, Bouchard J, Wynn SK, Mehta RL. Oliguria is an early predictor of higher mortality in critically ill patients. Kidney Int. 2011 Oct;80(7):760-7. doi: 10.1038/ki.2011.150. Epub 2011 Jun 29..
PMID: 23787055. Md Ralib A, Pickering JW, Shaw GM, Endre ZH. The urine output definition of acute kidney injury is too liberal. Crit Care. 2013 Jun 20;17(3):R112. doi: 10.1186/cc12784.
PMID: 29156029.Kunst G, Ostermann M. Intraoperative permissive oliguria – how much is too much? Br J Anaesth. 2017 Dec 1;119(6):1075-1077. doi: 10.1093/bja/aex387. Erratum in: Br J Anaesth. 2018 Mar;120(3):613.
PMID: 23760694. Finlay S, Bray B, Lewington AJ, Hunter-Rowe CT, Banerjee A, Atkinson JM, Jones MC. Identification of risk factors associated with acute kidney injury in patients admitted to acute medical units. Clin Med (Lond). 2013 Jun;13(3):233-8. doi: 10.7861/clinmedicine.13-3-233.
PMID: 18385668. Hsu CY, Ordoñez JD, Chertow GM, Fan D, McCulloch CE, Go AS. The risk of acute renal failure in patients with chronic kidney disease. Kidney Int. 2008 Jul;74(1):101-7. doi: 10.1038/ki.2008.107. Epub 2008 Apr 2.
PMID: 17088458. Mehta RH, Grab JD, O’Brien SM, Bridges CR, Gammie JS, Haan CK, Ferguson TB, Peterson ED; Society of Thoracic Surgeons National Cardiac Surgery Database Investigators. Bedside tool for predicting the risk of postoperative dialysis in patients undergoing cardiac surgery. Circulation. 2006 Nov 21;114(21):2208-16; quiz 2208. doi: 10.1161/CIRCULATIONAHA.106.635573. Epub 2006 Nov 6.
PMID: 15680458. Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005 Jan 29-Feb 4;365(9457):417-30. doi: 10.1016/S0140-6736(05)17831-3.
PMID: 27670788. Ostermann M, Joannidis M. Acute kidney injury 2016: diagnosis and diagnostic workup. Crit Care. 2016 Sep 27;20(1):299. doi: 10.1186/s13054-016-1478-z.
PMID: 35373012. Varghese V, Rivera MS, Alalwan AA, Alghamdi AM, Gonzalez ME, Velez JCQ. Diagnostic Utility of Serial Microscopic Examination of the Urinary Sediment in Acute Kidney Injury. Kidney360. 2020 Dec 11;2(2):182-191. doi: 10.34067/KID.0004022020.
PMID: 28545421. Manoeuvrier G, Bach-Ngohou K, Batard E, Masson D, Trewick D. Diagnostic performance of serum blood urea nitrogen to creatinine ratio for distinguishing prerenal from intrinsic acute kidney injury in the emergency department. BMC Nephrol. 2017 May 25;18(1):173. doi: 10.1186/s12882-017-0591-9.
PMID: 27236480. Legrand M, Le Cam B, Perbet S, Roger C, Darmon M, Guerci P, Ferry A, Maurel V, Soussi S, Constantin JM, Gayat E, Lefrant JY, Leone M; support of the AZUREA network. Urine sodium concentration to predict fluid responsiveness in oliguric ICU patients: a prospective multicenter observational study. Crit Care. 2016 May 29;20(1):165. doi: 10.1186/s13054-016-1343-0.
PMID: 24052222. Muriithi AK, Nasr SH, Leung N. Utility of urine eosinophils in the diagnosis of acute interstitial nephritis. Clin J Am Soc Nephrol. 2013 Nov;8(11):1857-62. doi: 10.2215/CJN.01330213. Epub 2013 Sep 19. Erratum in: Clin J Am Soc Nephrol. 2018 Jul 6;13(7):1079.
PMID: 28261499. Kaptein MJ, Kaptein EM. Focused Real-Time Ultrasonography for Nephrologists. Int J Nephrol. 2017;2017:3756857. doi: 10.1155/2017/3756857. Epub 2017 Feb 2.
PMID: 29866457. Lee SA, Cozzi M, Bush EL, Rabb H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am J Kidney Dis. 2018 Dec;72(6):846-856. doi: 10.1053/j.ajkd.2018.03.028. Epub 2018 Jun 14.
PMID: 25259720.Druml W. Systemic consequences of acute kidney injury. Curr Opin Crit Care. 2014 Dec;20(6):613-9. doi: 10.1097/MCC.0000000000000150.
PMID: 20027192.Prowle JR, Echeverri JE, Ligabo EV, Ronco C, Bellomo R. Fluid balance and acute kidney injury. Nat Rev Nephrol. 2010 Feb;6(2):107-15. doi: 10.1038/nrneph.2009.213. Epub 2009 Dec 22.
PMID: 32704211. Patil VP, Salunke BG. Fluid Overload and Acute Kidney Injury. Indian J Crit Care Med. 2020 Apr;24(Suppl 3):S94-S97. doi: 10.5005/jp-journals-10071-23401.
PMID: 30412310.Colbert GB, Szerlip HM. Euvolemia-A critical target in the management of acute kidney injury. Semin Dial. 2019 Jan;32(1):30-34. doi: 10.1111/sdi.12753. Epub 2018 Nov 9.
PMID: 34888157. Galindo P, Gasca C, Argaiz ER, Koratala A. Point of care venous Doppler ultrasound: Exploring the missing piece of bedside hemodynamic assessment. World J Crit Care Med. 2021 Nov 9;10(6):310-322. doi: 10.5492/wjccm.v10.i6.310.
PMID: 28114128. Ding X, Cheng Z, Qian Q. Intravenous Fluids and Acute Kidney Injury. Blood Purif. 2017;43(1-3):163-172. doi: 10.1159/000452702. Epub 2017 Jan 24.
PMID: 32704211.Patil VP, Salunke BG. Fluid Overload and Acute Kidney Injury. Indian J Crit Care Med. 2020 Apr;24(Suppl 3):S94-S97. doi: 10.5005/jp-journals-10071-23401.
PMID: 31589567.de Oliveira AM, Paulino MV, Vieira APF, McKinney AM, da Rocha AJ, Dos Santos GT, Leite CDC, Godoy LFS, Lucato LT. Imaging Patterns of Toxic and Metabolic Brain Disorders. Radiographics. 2019 Oct;39(6):1672-1695. doi: 10.1148/rg.2019190016.
PMID: 20615611. Kumar G, Goyal MK. Lentiform Fork sign: a unique MRI picture. Is metabolic acidosis responsible? Clin Neurol Neurosurg. 2010 Nov;112(9):805-12. doi: 10.1016/j.clineuro.2010.06.006. Epub 2010 Jul 7.
PMID: 22673882. Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012 Sep;82(5):516-24. doi: 10.1038/ki.2012.208. Epub 2012 Jun 6.
PMID: 26573630. Kato R, Pinsky MR. Personalizing blood pressure management in septic shock. Ann Intensive Care. 2015 Dec;5(1):41. doi: 10.1186/s13613-015-0085-5. Epub 2015 Nov 16.
PMID: 27230984. Ichai C, Vinsonneau C, Souweine B, Armando F, Canet E, Clec’h C, Constantin JM, Darmon M, Duranteau J, Gaillot T, Garnier A, Jacob L, Joannes-Boyau O, Juillard L, Journois D, Lautrette A, Muller L, Legrand M, Lerolle N, Rimmelé T, Rondeau E, Tamion F, Walrave Y, Velly L; Société française d’anesthésie et de réanimation (Sfar); Société de réanimation de langue française (SRLF); Groupe francophone de réanimation et urgences pédiatriques (GFRUP); Société française de néphrologie (SFN). Acute kidney injury in the perioperative period and in intensive care units (excluding renal replacement therapies). Ann Intensive Care. 2016 Dec;6(1):48. doi: 10.1186/s13613-016-0145-5. Epub 2016 May 27.
PMID: 27483065.Gordon AC, Mason AJ, Thirunavukkarasu N, Perkins GD, Cecconi M, Cepkova M, Pogson DG, Aya HD, Anjum A, Frazier GJ, Santhakumaran S, Ashby D, Brett SJ; VANISH Investigators. Effect of Early Vasopressin vs Norepinephrine on Kidney Failure in Patients With Septic Shock: The VANISH Randomized Clinical Trial. JAMA. 2016 Aug 2;316(5):509-18. doi: 10.1001/jama.2016.10485.
PMID: 20200382.De Backer D, Biston P, Devriendt J, Madl C, Chochrad D, Aldecoa C, Brasseur A, Defrance P, Gottignies P, Vincent JL; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010 Mar 4;362(9):779-89. doi: 10.1056/NEJMoa0907118.
PMID: 29485925. Semler MW, Self WH, Wanderer JP, Ehrenfeld JM, Wang L, Byrne DW, Stollings JL, Kumar AB, Hughes CG, Hernandez A, Guillamondegui OD, May AK, Weavind L, Casey JD, Siew ED, Shaw AD, Bernard GR, Rice TW; SMART Investigators and the Pragmatic Critical Care Research Group. Balanced Crystalloids versus Saline in Critically Ill Adults. N Engl J Med. 2018 Mar 1;378(9):829-839. doi: 10.1056/NEJMoa1711584. Epub 2018 Feb 27.
PMID: 29485926. Self WH, Semler MW, Wanderer JP, Wang L, Byrne DW, Collins SP, Slovis CM, Lindsell CJ, Ehrenfeld JM, Siew ED, Shaw AD, Bernard GR, Rice TW; SALT-ED Investigators. Balanced Crystalloids versus Saline in Noncritically Ill Adults. N Engl J Med. 2018 Mar 1;378(9):819-828. doi: 10.1056/NEJMoa1711586. Epub 2018 Feb 27.
PMID: 29485926. Self WH, Semler MW, Wanderer JP, Wang L, Byrne DW, Collins SP, Slovis CM, Lindsell CJ, Ehrenfeld JM, Siew ED, Shaw AD, Bernard GR, Rice TW; SALT-ED Investigators. Balanced Crystalloids versus Saline in Noncritically Ill Adults. N Engl J Med. 2018 Mar 1;378(9):819-828. doi: 10.1056/NEJMoa1711586. Epub 2018 Feb 27.
PMID: 29910040.Jaber S, Paugam C, Futier E, Lefrant JY, Lasocki S, Lescot T, Pottecher J, Demoule A, Ferrandière M, Asehnoune K, Dellamonica J, Velly L, Abback PS, de Jong A, Brunot V, Belafia F, Roquilly A, Chanques G, Muller L, Constantin JM, Bertet H, Klouche K, Molinari N, Jung B; BICAR-ICU Study Group. Sodium bicarbonate therapy for patients with severe metabolic acidaemia in the intensive care unit (BICAR-ICU): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet. 2018 Jul 7;392(10141):31-40. doi: 10.1016/S0140-6736(18)31080-8. Epub 2018 Jun 14. Erratum in: Lancet. 2018 Dec 8;392(10163):2440.
PMID: 11576874. Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis. 2001 Oct;38(4):703-27. doi: 10.1053/ajkd.2001.27688.
PMID: 22945490. Kraut JA, Madias NE. Treatment of acute metabolic acidosis: a pathophysiologic approach. Nat Rev Nephrol. 2012 Oct;8(10):589-601. doi: 10.1038/nrneph.2012.186. Epub 2012 Sep 4.
PMID: 22610181. RENAL Replacement Therapy Study Investigators, Bellomo R, Cass A, Cole L, Finfer S, Gallagher M, Lee J, Lo S, McArthur C, McGuiness S, Norton R, Myburgh J, Scheinkestel C, Su S. An observational study fluid balance and patient outcomes in the Randomized Evaluation of Normal vs. Augmented Level of Replacement Therapy trial. Crit Care Med. 2012 Jun;40(6):1753-60. doi: 10.1097/CCM.0b013e318246b9c6.
PMID: 25979272. Zhang L, Chen Z, Diao Y, Yang Y, Fu P. Associations of fluid overload with mortality and kidney recovery in patients with acute kidney injury: A systematic review and meta-analysis. J Crit Care. 2015 Aug;30(4):860.e7-13. doi: 10.1016/j.jcrc.2015.03.025. Epub 2015 Apr 9.
PMID: 29789983. Malbrain MLNG, Van Regenmortel N, Saugel B, De Tavernier B, Van Gaal PJ, Joannes-Boyau O, Teboul JL, Rice TW, Mythen M, Monnet X. Principles of fluid management and stewardship in septic shock: it is time to consider the four D’s and the four phases of fluid therapy. Ann Intensive Care. 2018 May 22;8(1):66. doi: 10.1186/s13613-018-0402-x.
PMID: 26798642. Benes J, Kirov M, Kuzkov V, Lainscak M, Molnar Z, Voga G, Monnet X. Fluid Therapy: Double-Edged Sword during Critical Care? Biomed Res Int. 2015;2015:729075. doi: 10.1155/2015/729075. Epub 2015 Dec 22.
PMID: 2967337. Lumlertgul N, Peerapornratana S, Trakarnvanich T, Pongsittisak W, Surasit K, Chuasuwan A, Tankee P, Tiranathanagul K, Praditpornsilpa K, Tungsanga K, Eiam-Ong S, Kellum JA, Srisawat N; FST Study Group. Early versus standard initiation of renal replacement therapy in furosemide stress test non-responsive acute kidney injury patients (the FST trial). Crit Care. 2018 Apr 19;22(1):101. doi: 10.1186/s13054-018-2021-1.
PMID: 25655065. Koyner JL, Davison DL, Brasha-Mitchell E, Chalikonda DM, Arthur JM, Shaw AD, Tumlin JA, Trevino SA, Bennett MR, Kimmel PL, Seneff MG, Chawla LS. Furosemide Stress Test and Biomarkers for the Prediction of AKI Severity. J Am Soc Nephrol. 2015 Aug;26(8):2023-31. doi: 10.1681/ASN.2014060535. Epub 2015 Feb 5.
PMID: 24053972.Chawla LS, Davison DL, Brasha-Mitchell E, Koyner JL, Arthur JM, Shaw AD, Tumlin JA, Trevino SA, Kimmel PL, Seneff MG. Development and standardization of a furosemide stress test to predict the severity of acute kidney injury. Crit Care. 2013 Sep 20;17(5):R207. doi: 10.1186/cc13015.
PMID: 31961246.Davenport MS, Perazella MA, Yee J, Dillman JR, Fine D, McDonald RJ, Rodby RA, Wang CL, Weinreb JC. Use of Intravenous Iodinated Contrast Media in Patients with Kidney Disease: Consensus Statements from the American College of Radiology and the National Kidney Foundation. Radiology. 2020 Mar;294(3):660-668. doi: 10.1148/radiol.2019192094. Epub 2020 Jan 21.
PMID: 32362431.Hinson JS, Ehmann MR, Klein EY. Evidence and Patient Safety Prevail Over Myth and Dogma: Consensus Guidelines on the Use of Intravenous Contrast Media. Ann Emerg Med. 2020 Aug;76(2):149-152. doi: 10.1016/j.annemergmed.2020.03.022. Epub 2020 Apr 30.
PMID: 31961246.Davenport MS, Perazella MA, Yee J, Dillman JR, Fine D, McDonald RJ, Rodby RA, Wang CL, Weinreb JC. Use of Intravenous Iodinated Contrast Media in Patients with Kidney Disease: Consensus Statements from the American College of Radiology and the National Kidney Foundation. Radiology. 2020 Mar;294(3):660-668. doi: 10.1148/radiol.2019192094. Epub 2020 Jan 21.
PMID: 28540198. Ozkok S, Ozkok A. Contrast-induced acute kidney injury: A review of practical points. World J Nephrol. 2017 May 6;6(3):86-99. doi: 10.5527/wjn.v6.i3.86.
PMID: 15680458. Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005 Jan 29-Feb 4;365(9457):417-30. doi: 10.1016/S0140-6736(05)17831-3.
PMID: 32668114.STARRT-AKI Investigators; Canadian Critical Care Trials Group; Australian and New Zealand Intensive Care Society Clinical Trials Group; United Kingdom Critical Care Research Group; Canadian Nephrology Trials Network; Irish Critical Care Trials Group, Bagshaw SM, Wald R, Adhikari NKJ, Bellomo R, da Costa BR, Dreyfuss D, Du B, Gallagher MP, Gaudry S, Hoste EA, Lamontagne F, Joannidis M, Landoni G, Liu KD, McAuley DF, McGuinness SP, Neyra JA, Nichol AD, Ostermann M, Palevsky PM, Pettilä V, Quenot JP, Qiu H, Rochwerg B, Schneider AG, Smith OM, Thomé F, Thorpe KE, Vaara S, Weir M, Wang AY, Young P, Zarbock A. Timing of Initiation of Renal-Replacement Therapy in Acute Kidney Injury. N Engl J Med. 2020 Jul 16;383(3):240-251. doi: 10.1056/NEJMoa2000741. Erratum in: N Engl J Med. 2020 Jul 15.
PMID: 10729745.Holt S, Moore K. Pathogenesis of renal failure in rhabdomyolysis: the role of myoglobin. Exp Nephrol. 2000 Mar-Apr;8(2):72-6. doi: 10.1159/000020651.
PMID: 10906171.Vanholder R, Sever MS, Erek E, Lameire N. Rhabdomyolysis. J Am Soc Nephrol. 2000 Aug;11(8):1553-1561. doi: 10.1681/ASN.V1181553.
PMID: 15774072. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: Rhabdomyolysis — an overview for clinicians. Crit Care. 2005 Apr;9(2):158-69. doi: 10.1186/cc2978. Epub 2004 Oct 20.
PMID: 10906171.Vanholder R, Sever MS, Erek E, Lameire N. Rhabdomyolysis. J Am Soc Nephrol. 2000 Aug;11(8):1553-1561. doi: 10.1681/ASN.V1181553.
PMID: 32532456.Cabral BMI, Edding SN, Portocarrero JP, Lerma EV. Rhabdomyolysis. Dis Mon. 2020 Aug;66(8):101015. doi: 10.1016/j.disamonth.2020.101015. Epub 2020 Jun 10.
PMID: 27259093.Simpson JP, Taylor A, Sudhan N, Menon DK, Lavinio A. Rhabdomyolysis and acute kidney injury: creatine kinase as a prognostic marker and validation of the McMahon Score in a 10-year cohort: A retrospective observational evaluation. Eur J Anaesthesiol. 2016 Dec;33(12):906-912. doi: 10.1097/EJA.0000000000000490.
PMID: 15774072. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: Rhabdomyolysis — an overview for clinicians. Crit Care. 2005 Apr;9(2):158-69. doi: 10.1186/cc2978. Epub 2004 Oct 20.
PMID: 27301374. Chavez LO, Leon M, Einav S, Varon J. Beyond muscle destruction: a systematic review of rhabdomyolysis for clinical practice. Crit Care. 2016 Jun 15;20(1):135. doi: 10.1186/s13054-016-1314-5.
PMID: 18311727.Kellum JA, Cerda J, Kaplan LJ, Nadim MK, Palevsky PM. Fluids for prevention and management of acute kidney injury. Int J Artif Organs. 2008 Feb;31(2):96-110. doi: 10.1177/039139880803100204.
PMID: 18311727.Kellum JA, Cerda J, Kaplan LJ, Nadim MK, Palevsky PM. Fluids for prevention and management of acute kidney injury. Int J Artif Organs. 2008 Feb;31(2):96-110. doi: 10.1177/039139880803100204.
PMID: 31774551.Durani U, Hogan WJ. Emergencies in haematology: tumour lysis syndrome. Br J Haematol. 2020 Feb;188(4):494-500. doi: 10.1111/bjh.16278. Epub 2019 Nov 27.
PMID: 29230244. Belay Y, Yirdaw K, Enawgaw B. Tumor Lysis Syndrome in Patients with Hematological Malignancies. J Oncol. 2017;2017:9684909. doi: 10.1155/2017/9684909. Epub 2017 Nov 2.
PMID: 30499695.Williams SM, Killeen AA. Tumor Lysis Syndrome. Arch Pathol Lab Med. 2019 Mar;143(3):386-393. doi: 10.5858/arpa.2017-0278-RS. Epub 2018 Nov 30.
PMID: 29230244. Belay Y, Yirdaw K, Enawgaw B. Tumor Lysis Syndrome in Patients with Hematological Malignancies. J Oncol. 2017;2017:9684909. doi: 10.1155/2017/9684909. Epub 2017 Nov 2.
PMID: 24359983.Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014 Jan;21(1):18-26. doi: 10.1053/j.ackd.2013.07.001.
PMID: 18509186.Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008 Jun 1;26(16):2767-78. doi: 10.1200/JCO.2007.15.0177. Erratum in: J Clin Oncol. 2010 Feb 1;28(4):708.
PMID: 25938028. Mirrakhimov AE, Voore P, Khan M, Ali AM. Tumor lysis syndrome: A clinical review. World J Crit Care Med. 2015 May 4;4(2):130-8. doi: 10.5492/wjccm.v4.i2.130.
PMID: 27888526. Yu X, Liu L, Nie X, Li J, Zhang J, Zhao L, Wang X. The optimal single-dose regimen of rasburicase for management of tumour lysis syndrome in children and adults: a systematic review and meta-analysis. J Clin Pharm Ther. 2017 Feb;42(1):18-26. doi: 10.1111/jcpt.12479. Epub 2016 Nov 25.
PMID: 24359983. Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014 Jan;21(1):18-26. doi: 10.1053/j.ackd.2013.07.001.
PMID: 25876990.Jones GL, Will A, Jackson GH, Webb NJ, Rule S; British Committee for Standards in Haematology. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015 Jun;169(5):661-71. doi: 10.1111/bjh.13403. Epub 2015 Apr 15.
PMID: 21561350. Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011 May 12;364(19):1844-54. doi: 10.1056/NEJMra0904569. Erratum in: N Engl J Med. 2018 Sep 13;379(11):1094.
PMID: 12799335.Balogh Z, McKinley BA, Cocanour CS, Kozar RA, Valdivia A, Sailors RM, Moore FA. Supranormal trauma resuscitation causes more cases of abdominal compartment syndrome. Arch Surg. 2003 Jun;138(6):637-42; discussion 642-3. doi: 10.1001/archsurg.138.6.637.
PMID: 32832400. Tariq R, Singal AK. Management of Hepatorenal Syndrome: A Review. J Clin Transl Hepatol. 2020 Jun 28;8(2):192-199. doi: 10.14218/JCTH.2020.00011. Epub 2020 Jun 1.
PMID: 27339675. Regner KR, Singbartl K. Kidney Injury in Liver Disease. Crit Care Clin. 2016 Jul;32(3):343-55. doi: 10.1016/j.ccc.2016.03.005.
PMID: 31302175.Angeli P, Garcia-Tsao G, Nadim MK, Parikh CR. News in pathophysiology, definition and classification of hepatorenal syndrome: A step beyond the International Club of Ascites (ICA) consensus document. J Hepatol. 2019 Oct;71(4):811-822. doi: 10.1016/j.jhep.2019.07.002. Epub 2019 Jul 11.
PMID: 34675586. Chaney A. A Review for the Practicing Clinician: Hepatorenal Syndrome, a Form of Acute Kidney Injury, in Patients with Cirrhosis. Clin Exp Gastroenterol. 2021 Oct 5;14:385-396. doi: 10.2147/CEG.S323778.
PMID: 32928750.Simonetto DA, Gines P, Kamath PS. Hepatorenal syndrome: pathophysiology, diagnosis, and management. BMJ. 2020 Sep 14;370:m2687. doi: 10.1136/bmj.m2687.
PMID: 8514039.Ginès A, Escorsell A, Ginès P, Saló J, Jiménez W, Inglada L, Navasa M, Clària J, Rimola A, Arroyo V, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993 Jul;105(1):229-36. doi: 10.1016/0016-5085(93)90031-7.
PMID: 29532591. Wong F, Jepsen P, Watson H, Vilstrup H. Un-precipitated acute kidney injury is uncommon among stable patients with cirrhosis and ascites. Liver Int. 2018 Oct;38(10):1785-1792. doi: 10.1111/liv.13738. Epub 2018 Mar 31.
PMID: 10432325.Sort P, Navasa M, Arroyo V, Aldeguer X, Planas R, Ruiz-del-Arbol L, Castells L, Vargas V, Soriano G, Guevara M, Ginès P, Rodés J. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999 Aug 5;341(6):403-9. doi: 10.1056/NEJM199908053410603.
PMID: 17854593.Fernández J, Navasa M, Planas R, Montoliu S, Monfort D, Soriano G, Vila C, Pardo A, Quintero E, Vargas V, Such J, Ginès P, Arroyo V. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007 Sep;133(3):818-24. doi: 10.1053/j.gastro.2007.06.065. Epub 2007 Jul 3.
PMID: 17235705. Esrailian E, Pantangco ER, Kyulo NL, Hu KQ, Runyon BA. Octreotide/Midodrine therapy significantly improves renal function and 30-day survival in patients with type 1 hepatorenal syndrome. Dig Dis Sci. 2007 Mar;52(3):742-8. doi: 10.1007/s10620-006-9312-0.
PMID: 19776409.Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009 Sep 24;361(13):1279-90. doi: 10.1056/NEJMra0809139. Erratum in: N Engl J Med. 2011 Jan 27;364(4):389.
PMID: 31723234.Velez JCQ, Therapondos G, Juncos LA. Reappraising the spectrum of AKI and hepatorenal syndrome in patients with cirrhosis. Nat Rev Nephrol. 2020 Mar;16(3):137-155. doi: 10.1038/s41581-019-0218-4. Epub 2019 Nov 13. Erratum in: Nat Rev Nephrol. 2020 Jan 27.
PMID: 25638527.Angeli P, Ginès P, Wong F, Bernardi M, Boyer TD, Gerbes A, Moreau R, Jalan R, Sarin SK, Piano S, Moore K, Lee SS, Durand F, Salerno F, Caraceni P, Kim WR, Arroyo V, Garcia-Tsao G. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. J Hepatol. 2015 Apr;62(4):968-74. doi: 10.1016/j.jhep.2014.12.029. Epub 2015 Jan 28. Erratum in: J Hepatol. 2015 Jul;63(1):290.
PMID: 32846202.Ojeda-Yuren AS, Cerda-Reyes E, Herrero-Maceda MR, Castro-Narro G, Piano S. An Integrated Review of the Hepatorenal Syndrome. Ann Hepatol. 2021 May-Jun;22:100236. doi: 10.1016/j.aohep.2020.07.008. Epub 2020 Aug 23.
PMID: 25638527. Angeli P, Ginès P, Wong F, Bernardi M, Boyer TD, Gerbes A, Moreau R, Jalan R, Sarin SK, Piano S, Moore K, Lee SS, Durand F, Salerno F, Caraceni P, Kim WR, Arroyo V, Garcia-Tsao G. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. J Hepatol. 2015 Apr;62(4):968-74. doi: 10.1016/j.jhep.2014.12.029. Epub 2015 Jan 28. Erratum in: J Hepatol. 2015 Jul;63(1):290.
PMID: 34675586. Chaney A. A Review for the Practicing Clinician: Hepatorenal Syndrome, a Form of Acute Kidney Injury, in Patients with Cirrhosis. Clin Exp Gastroenterol. 2021 Oct 5;14:385-396. doi: 10.2147/CEG.S323778.
PMID: 24076364.Ge PS, Runyon BA. The changing role of beta-blocker therapy in patients with cirrhosis. J Hepatol. 2014 Mar;60(3):643-53. doi: 10.1016/j.jhep.2013.09.016. Epub 2013 Sep 26.
PMID: 32928750. Simonetto DA, Gines P, Kamath PS. Hepatorenal syndrome: pathophysiology, diagnosis, and management. BMJ. 2020 Sep 14;370:m2687. doi: 10.1136/bmj.m2687.
PMID: 29653741.European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu; European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018 Aug;69(2):406-460. doi: 10.1016/j.jhep.2018.03.024. Epub 2018 Apr 10. Erratum in: J Hepatol. 2018 Nov;69(5):1207.
PMID: 31525507. Paugam-Burtz C, Levesque E, Louvet A, Thabut D, Amathieu R, Bureau C, Camus C, Chanques G, Faure S, Ferrandière M, Francoz C, Galbois A, Gustot T, Ichai C, Ichai P, Jaber S, Lescot T, Moreau R, Roullet S, Saliba F, Thévenot T, Velly L, Weiss E. Management of liver failure in general intensive care unit. Anaesth Crit Care Pain Med. 2020 Feb;39(1):143-161. doi: 10.1016/j.accpm.2019.06.014. Epub 2019 Sep 13.
PMID: 28991106.Nanda A, Reddy R, Safraz H, Salameh H, Singal AK. Pharmacological Therapies for Hepatorenal Syndrome: A Systematic Review and Meta-Analysis. J Clin Gastroenterol. 2018 Apr;52(4):360-367. doi: 10.1097/MCG.0000000000000913.
PMID: 19885875.Gluud LL, Christensen K, Christensen E, Krag A. Systematic review of randomized trials on vasoconstrictor drugs for hepatorenal syndrome. Hepatology. 2010 Feb;51(2):576-84. doi: 10.1002/hep.23286.
PMID: 28403995.Facciorusso A, Chandar AK, Murad MH, Prokop LJ, Muscatiello N, Kamath PS, Singh S. Comparative efficacy of pharmacological strategies for management of type 1 hepatorenal syndrome: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2017 Feb;2(2):94-102. doi: 10.1016/S2468-1253(16)30157-1. Epub 2016 Dec 2.
PMID: 29653741.European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu; European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018 Aug;69(2):406-460. doi: 10.1016/j.jhep.2018.03.024. Epub 2018 Apr 10. Erratum in: J Hepatol. 2018 Nov;69(5):1207.
PMID: 28953318. Israelsen M, Krag A, Allegretti AS, Jovani M, Goldin AH, Winter RW, Gluud LL. Terlipressin versus other vasoactive drugs for hepatorenal syndrome. Cochrane Database Syst Rev. 2017 Sep 27;9(9):CD011532. doi: 10.1002/14651858.CD011532.pub2.
PMID: 32928750.Simonetto DA, Gines P, Kamath PS. Hepatorenal syndrome: pathophysiology, diagnosis, and management. BMJ. 2020 Sep 14;370:m2687. doi: 10.1136/bmj.m2687.
PMID: 32928750. Simonetto DA, Gines P, Kamath PS. Hepatorenal syndrome: pathophysiology, diagnosis, and management. BMJ. 2020 Sep 14;370:m2687. doi: 10.1136/bmj.m2687.
PMID: 28991106.Nanda A, Reddy R, Safraz H, Salameh H, Singal AK. Pharmacological Therapies for Hepatorenal Syndrome: A Systematic Review and Meta-Analysis. J Clin Gastroenterol. 2018 Apr;52(4):360-367. doi: 10.1097/MCG.0000000000000913.
PMID: 15956066.Kiser TH, Fish DN, Obritsch MD, Jung R, MacLaren R, Parikh CR. Vasopressin, not octreotide, may be beneficial in the treatment of hepatorenal syndrome: a retrospective study. Nephrol Dial Transplant. 2005 Sep;20(9):1813-20. doi: 10.1093/ndt/gfh930. Epub 2005 Jun 14.
PMID: 25644760.Cavallin M, Kamath PS, Merli M, Fasolato S, Toniutto P, Salerno F, Bernardi M, Romanelli RG, Colletta C, Salinas F, Di Giacomo A, Ridola L, Fornasiere E, Caraceni P, Morando F, Piano S, Gatta A, Angeli P; Italian Association for the Study of the Liver Study Group on Hepatorenal Syndrome. Terlipressin plus albumin versus midodrine and octreotide plus albumin in the treatment of hepatorenal syndrome: A randomized trial. Hepatology. 2015 Aug;62(2):567-74. doi: 10.1002/hep.27709. Epub 2015 Feb 13.
PMID: 12830007.Pomier-Layrargues G, Paquin SC, Hassoun Z, Lafortune M, Tran A. Octreotide in hepatorenal syndrome: a randomized, double-blind, placebo-controlled, crossover study. Hepatology. 2003 Jul;38(1):238-43. doi: 10.1053/jhep.2003.50276.
PMID: 24751830.Arroyo V, García-Martinez R, Salvatella X. Human serum albumin, systemic inflammation, and cirrhosis. J Hepatol. 2014 Aug;61(2):396-407. doi: 10.1016/j.jhep.2014.04.012. Epub 2014 Apr 18.
PMID: 26606982. Salerno F, Navickis RJ, Wilkes MM. Albumin treatment regimen for type 1 hepatorenal syndrome: a dose-response meta-analysis. BMC Gastroenterol. 2015 Nov 25;15:167. doi: 10.1186/s12876-015-0389-9.
PMID: 32572647. Aldecoa C, Llau JV, Nuvials X, Artigas A. Role of albumin in the preservation of endothelial glycocalyx integrity and the microcirculation: a review. Ann Intensive Care. 2020 Jun 22;10(1):85. doi: 10.1186/s13613-020-00697-1.
Add comment