

20 November 2024, by Shahriar Lahouti.

CONTENTS
- Preface
- Meaning of Acid-Base
- Acid-base balance
- Approach to Acid-Base Disorders
- Metabolic acidosis
- Metabolic alkalosis
- Respiratory Acid-base Disorders
- Case Vignette
- Going further
- Appendix
- References
Preface
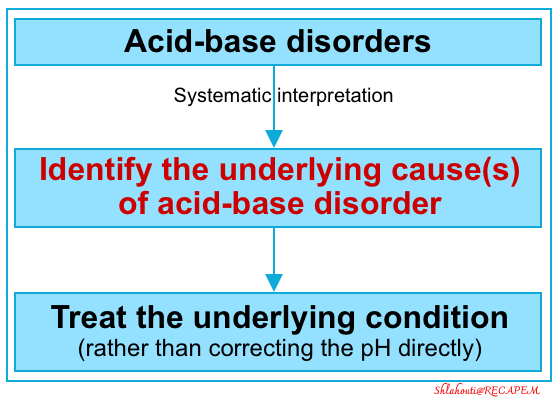
Acid-base disorders are a common occurrence in critically ill patients, and it is crucial to approach them systematically; focusing on determining the underlying cause of abnormalities and intervening when appropriate. Serum pH alone neither defines a specific disease nor specifies an appropriate therapy. Therefore, the actual pH matters far less than the underlying cause of the acid-base abnormality. Treatment intended to correct laboratory values alone (ie, serum pH) is not helpful and can sometimes be detrimental. Instead, the underlying cause of the abnormality must be assessed, with the results of the assessment providing the key to resuscitation.
| Case scenario A 24-year-old man with a history of type 1 DM presented to the hospital for the 7-hour duration of abdominal pain and vomiting. Current medication includes NPH insulin of 17 units daily. He had a significant history of non-adherence with his insulin regimen causing the precipitation of the symptoms. BP was 124/86 mm Hg, HR 83/minute, afebrile, RR 22/minute, and pulse oximetry 98% on ambient air. The physical examination was positive for epigastric tenderness, dry mucous membranes, and decreased skin turgor. Labs: Na 130 meq/L, K 3.6 meq/L, Cl 88 meq/L, HCO3 21 meq/L, serum glucose 494 mg/dL, albumin 5.9 g/L, creatinine 1.25 mg/dL, lactate 0.6 mmom/L and highly positive serum ketones. Venous blood gas: pH 7.45, PCO2 28 mmHg. How do we interpret his blood gases? |
Meaning of Acid-Base
◾️Background
- Serum pH is a measurement of the activity of free protons in the plasma (pH = − log10[H+]).
-
A pH value of 7.4 corresponds to a H+ concentration of ~40 nmol/L.
-
- Acid-base disorder historically refers to the alteration of pH out of physiologic range.
◾️Definition
- It is important to differentiate acidemia from acidosis, and alkalemia from alkalosis.
-
Acidemia, vs. acidosis
-
Acidemia is a direct measure of the serum H+ concentration; when pH is < 7.35 this is called acidemia.
-
Acidosis: This is defined as the clinical processes that tend to lower the pH to <7.35. A patient can have multiple acidoses or alkaloses that, in sum, give rise to the serum pH.
-
-
Alkalemia vs. alkalosis
- Alkalemia is a direct measure of the serum H+ concentration; when pH is > 7.45 this is called alkalemia.
- Alkalosis is defined as the clinical processes that tend to increase serum alkalinity. A patient can have multiple acidoses or alkaloses that, in sum, give rise to the serum pH.
-
◾️Clinical significance of pH values
- The adverse consequences of acidemia and alkalemia are listed below.
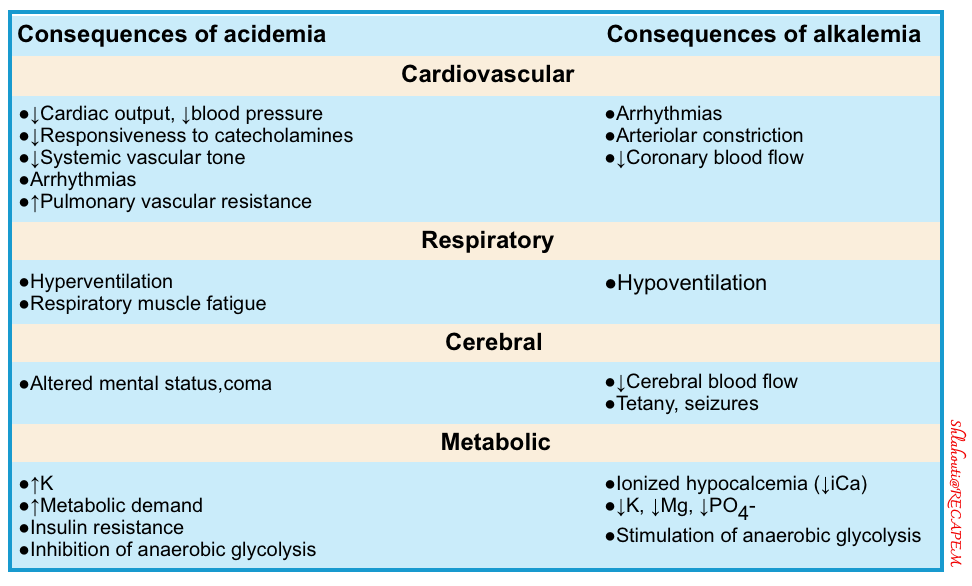
- However, the pH number matters far less than the underlying cause of the acid-base abnormality. Any acid-base schema should prompt one to identify the causative factors relating to the acid-base perturbations and then strive to correct these rather than directly correct the pH.
- It is a commonly held belief that acidemia negatively affects a patient’s cardiac function and response to vasopressor agents.
- This dogma is based on animal models that have never been validated in human studies. In contrast, several clinical trials have demonstrated that correcting the serum pH without intervening in the underlying problem does not correct the hemodynamic dysfunction. Instead, likely, the cause of the cardiac dysfunction and vasopressor resistance in the animal model studies was the underlying disease leading to the acidosis in question.
- A significant body of evidence suggests that acidosis in itself causes only minimal physiologic unrest.
- Frumin demonstrated patients’ ability to tolerate acidosis from hypercapnia to extremely low levels (pH as low as 6.8) without any signs of physiologic distress or hemodynamic compromise. For example, shortly after a race, Olympic rowers have exhibited serum pH values as low as 6.8 without any ill effects.
- Patients presenting with diabetic ketoacidosis (DKA) with impressively severe acidosis rarely demonstrate hemodynamic compromise. In contrast, the septic patient with lactic acidosis and a pH of 7.1 is often in refractory shock and dying *, *.
- Multiple studies have demonstrated that the prognosis of patients with acidosis is not related to serum pH but rather to the underlying cause of the acid-base abnormality *, *, *, *.
- It is a commonly held belief that acidemia negatively affects a patient’s cardiac function and response to vasopressor agents.
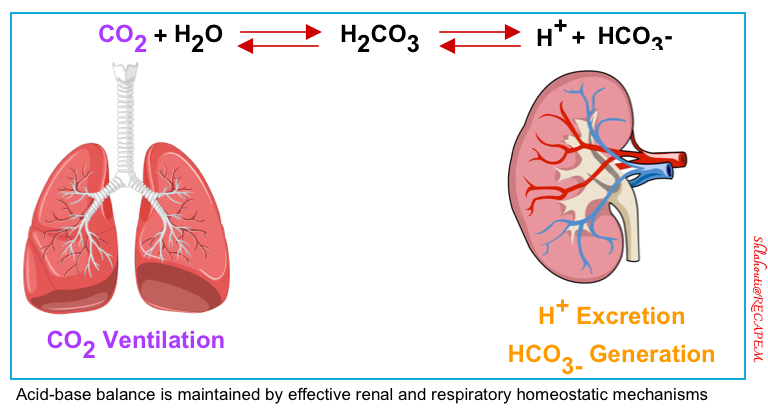
Acid-Base balance
- A buffer is a solution that resists a change in pH. The key buffer is the bicarbonate system present in the extracellular fluid. Like any buffer, this system comprises a weak acid (in this case carbonic acid, H2CO3) and its conjugate base (the bicarbonate ion, HCO3–), which exist in dynamic equilibrium as shown in the following equation.
- The traditional approach is based on the Henderson–Hasselbalch equation (figure below), which states that H+ concentration is proportional to PCO2/HCO3−
- This approach suggests that a primary change in the PaCO2 will cause secondary changes in bicarbonate and vice versa (also known as an “adaptive” response).
- Based on this assumption, PCO2 and HCO3– are independent variables.
- This new approach doesn’t deny the traditional approach, rather it may provide the overall and bigger picture.
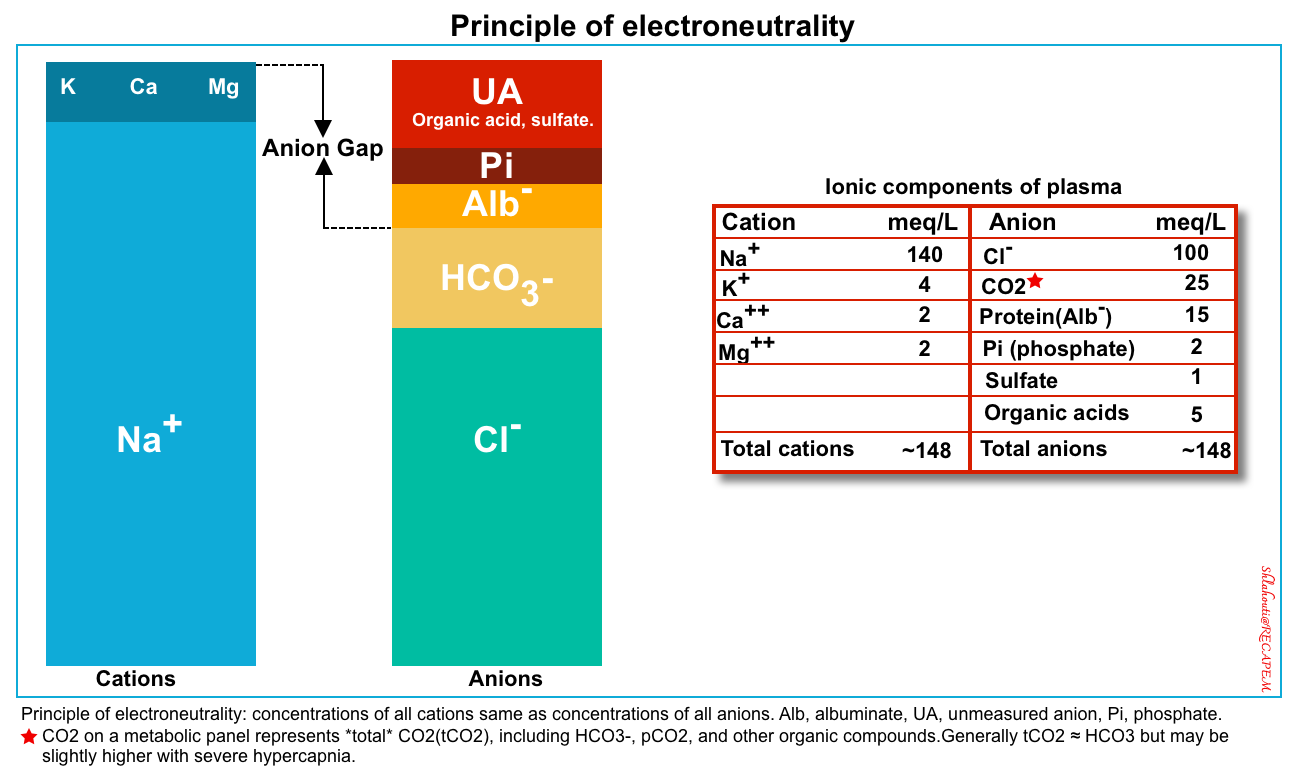
- The key concept of the new Stewart approach is electroneutrality.
- The principle of electroneutrality implies that the concentration of all plasma cations should be equal to the concentration of all anions to maintain the electrical equilibrium as shown in the figure below *.
-
Cations: Na+, K+, Ca2+, Mg2+ (for practical purposes, use only the Na value because this is the major cation present in the serum).
-
Anions: Cl−, HCO3−, and the remainder, which is typically referred to as the “anion gap“.
-
A normal anion gap is filled by weak acids {albuminate (Alb–), phosphate(Pi)}, and unmeasured anions (i.e. organic acid, and some inorganic acids).
- Reminder: Endogenous acids include:
- Volatile acid (CO2) that is eliminated by the lungs.
- Organic acids: Primarily ketones & lactate which are eliminated by the liver.
- Inorganic acids: Primarily sulfate and phosphate that are eliminated by the kidney.
-
-
- The principle of electroneutrality implies that the concentration of all plasma cations should be equal to the concentration of all anions to maintain the electrical equilibrium as shown in the figure below *.
- One of the key concepts of the new Stewart approach is that bicarbonate, or HCO3− does not play any role in acid-base balance as opposed to the traditional. approach.
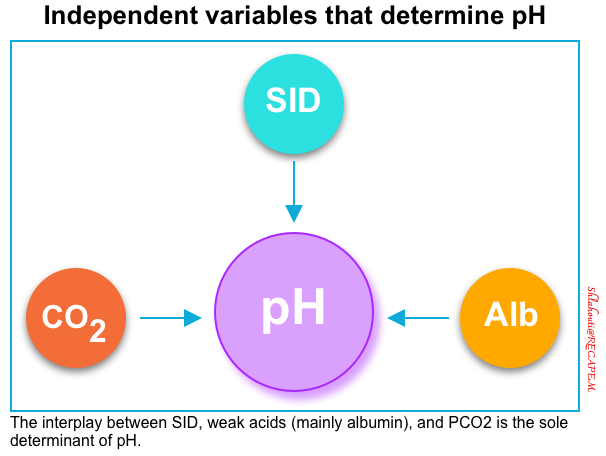
- According to the Stewart approach, there are only three independent variables that determine the concentration of H+ and thus pH in any fluid *:
- Strong Ion Difference (SID)
- Weak acids (mainly albumin, and phosphate)
- Partial pressure of carbon dioxide (PCO2)
- Thus, while HCO3− may follow the change in one of these independent variables, it can never cause a change in the pH by itself.
Strong ion difference (SID)
- SID is a major determinant of acid-base status and pH.
- Strong ions completely dissociate in water and do not remain in their molecular forms (i.e. NaCl is not in solution as molecules of NaCl, but rather as ions of Na+ and Cl−).
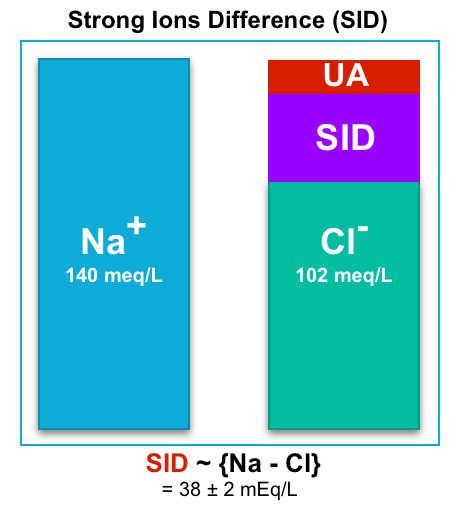
- Strong ion difference is the sum of all positively charged fully dissociated ions (mainly Na+) minus the sum of all negatively charged fully dissociated ions (mainly Cl−).
- A simplified “SID” equation is restricted only to {Na – Cl} *.
- A simplified “SID” equation is restricted only to {Na – Cl} *.
- The normal value for SID is ~ 38 (38 ± 2 mEq/L).
- A “SID” > 40 implies that the body has excess cations. To maintain electroneutrality, the body should generate an anion (e.g. HCO3–).
- A “SID” < 36 implies that the body has cation deficiency. So to maintain electroneutrality, the body should generate cation (i.e. H+).
- In the physiologic state, the gap between strong cations and anions (i.e. SID) is filled up mostly by HCO3 and weak anions comprising mostly of Alb-. See the figure above.
- Thus, SID can also be calculated alternatively as the sum of HCO3− and weak anions.
Weak Acids (anions): Albumin
- Albumin is a weak acid (weak acids only partially dissociate in water).
- The amount of H+ locked on albumin depends on body pH *.
- Albumin molecules have many positive and negative charges due to amino acid side chains that release or bind hydrogen ions. The ratio of albumin’s positive and negative charges changes with the pH of the solution.
- An increase in weak acids (hyperalbuminemia), which occurs mainly in dehydrated conditions, can result in metabolic acidosis and a decrease in HCO3− (with unchanged SID) *.
- Similarly, a reduction of weak acids from reduced production (liver failure) or increased loss (nephrotic syndrome) results in mild metabolic alkalosis and an increase in HCO3− *.
Total CO2
- CO2 is a respiratory determinant of acid-base status.
-
CO2, while not an acid itself, functions as one through the formation of carbonic acid.
- The body tightly regulates CO2 hemostasis by modifying respiratory drive and minute ventilation to maintain pCO2 hemostasis closely.
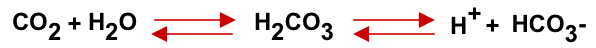
- Stewart’s approach emphasizes [H2CO3]/[HCO3−] equilibrium. But maintains that ultimately what counts is the total CO2, as long as there is sufficient carbonic anhydrase (an enzyme that modifies the reaction between H2O and CO2 generating HCO3− and H+), intact circulation (that carries CO2 from tissue to lung), and normal functioning lungs (that regulate PaCO2).
Approach to Acid-Base Disorders
Systematic Approach
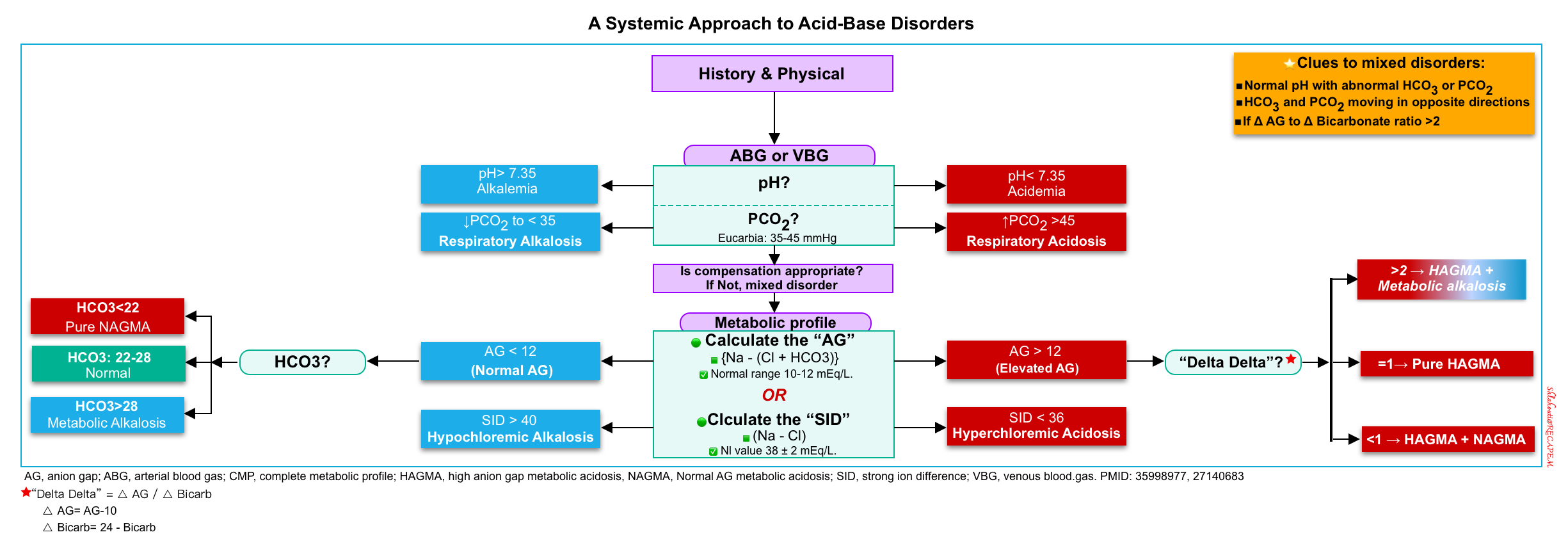
| Acid-Base Interpretation Steps |
- History and physical exam.
- Acidemia or alkalemia? Primary disturbance?
- Observe the pH, PaCO2 and HCO3
- Keep in mind that a pH value within the normal range does not rule out existing acid-base disturbances.
- The purpose of this step is to look for any acid-base abnormality and to recognize a primary disorder (if any).
- Remember that the so-called primary disorder is solely responsible for the purpose of calculating compensatory response and not to give undue importance to one disorder over another.
- Observe the pH, PaCO2 and HCO3
- Look for the compensatory response.
- Compensatory responses are calculated based on empirical formulae to identify any second disorder. See here.
- Note that, the body does not overcompensate.
- Compensatory responses are calculated based on empirical formulae to identify any second disorder. See here.
- Calculate the anion gap. MDCalc 🧮
- The presence of high AG may be the sole pointer towards the presence of hidden metabolic acidosis, with multiple opposing acid-base abnormalities normalizing pH, PaCO2, and HCO3.
- Delta gap? Calculator 🧮
- SID? see here.
- Single vs. multiple acid-base disorder?
- Mixed acid-base disorders are common in critically ill patients such as salicylate intoxication, sepsis, liver failure *, etc.
- How does this explain the clinical picture?
History and Physical
◾️Background
- The accuracy of historical and physical exam findings is insufficient for the independent identification of acid-base disorders.
- However, since a patient’s pH is, in itself, minimally significant, clinicians should instead use the patient’s history and physical exam to elucidate the etiology of the patient’s presentation.
- A patient’s acid-base status, whether respiratory or metabolic, can only be evaluated within that patient’s clinical context. For example, a patient suffering from chronic hypercapnia requires no active intervention *. As such, blood gas analysis rarely affects patient management in the hyperacute setting (see below).
◾️History: Clues in the history include *
- Certain diseases are known to be associated with acid-base disorders, for example *
- Vomiting (metabolic alkalosis)
- Diarrhea (metabolic acidosis)
- Chronic obstructive pulmonary disease (respiratory acidosis)
- Pneumonia (respiratory alkalosis)
- Medication list 💊
- Diuretics (see appendix).
- Paracetamol, salicylates?
- Opioids? (respiratory depressants)
- Ingestions, e.g. alcohols?
- Comorbidities, e.g. COPD, DM, liver disease, chronic kidney disease?
- Archival (baseline) blood gas and chemistry?
🔳Physicals 🩺
- Check the patient’s blood pressure, heart rate, and respiratory rate.
- Neurologic state: Check if the patient is conscious or unconscious, neuromuscular weaknesses?
- Signs of infection: Check for fever.
- Pulmonary status: Check for Kussmaul respiration (pattern of breathing, more on this, here 📖), cyanosis, and clubbing of the fingers.
Blood gas analysis

◾️Clinical significance- What does the blood gas analysis add to a clinically relevant pH analysis?
- It provides information regarding respiratory pH abnormalities (i.e. PCO2).
- Blood gas analysis usually will not have a major effect on the diagnosis and management of the patient (assuming that the electrolyte panel is evaluated and a thoughtful history and physical examination is performed).
- Blood gas analysis can answer two questions:
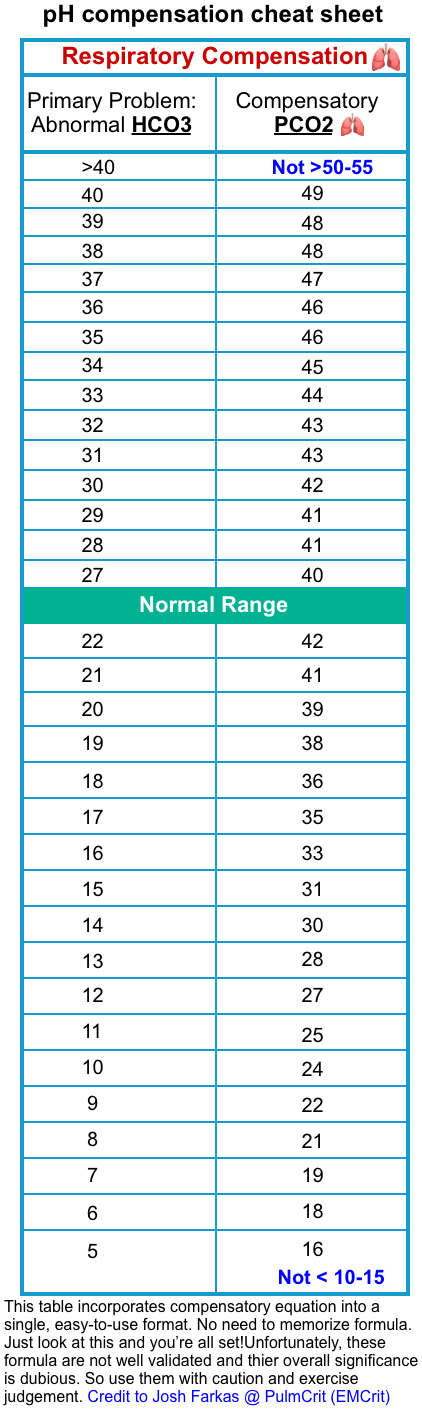
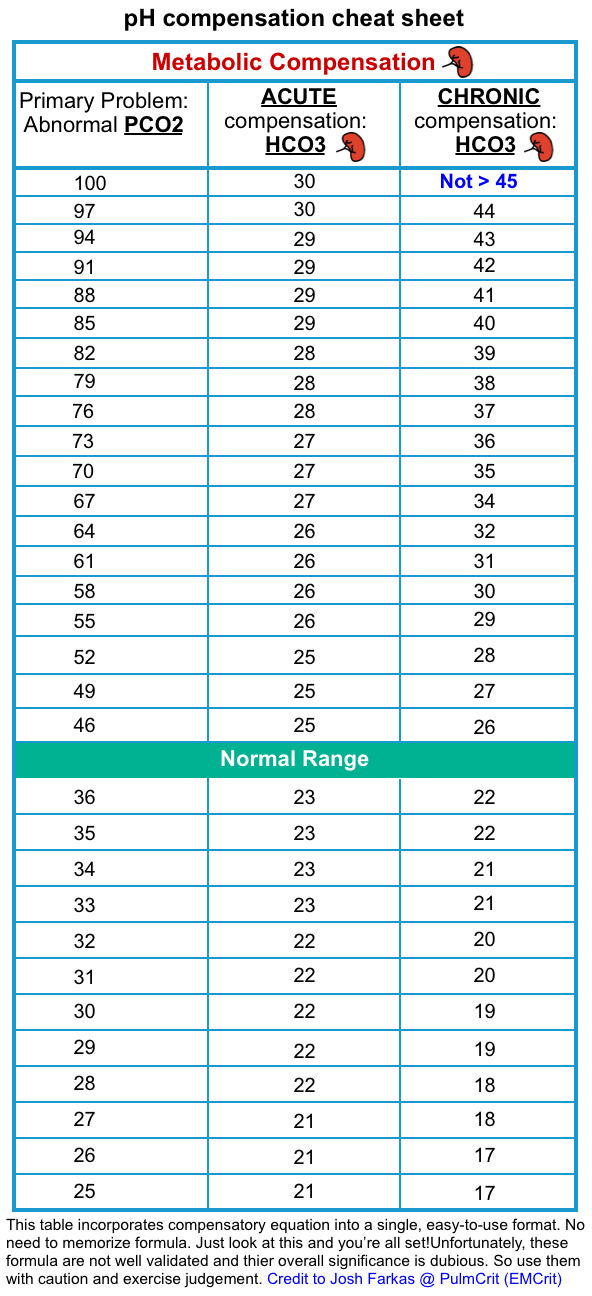
- Is there adequate respiratory compensation?
- Patients with metabolic pH abnormalities should “normally” be expected to develop respiratory compensation as:
- Metabolic alkalosis may be compensated for by mild hypoventilation (secondary respiratory acidosis)
- Metabolic acidosis may be compensated for by hyperventilation (secondary respiratory alkalosis)
- By identifying whether the patient is following these rules, we could theoretically determine whether their ventilation is intact (or whether it is proportionally inadequate or excessive). However, several caveats should be mentioned here:
- In critically ill patients, ventilation is influenced by numerous factors (e.g. anxiety, opioids, mechanical ventilation settings). Compensation equations (Winter’s Formula) were derived from a cohort of stable patients without multiple active problems.
- According to modern clinical practice, decisions regarding intubation or the selection of respiratory support devices are generally made based on clinical assessment (e.g. work of breathing, mental status, etc.) and the diagnosis, but not the blood gas values by itself.
- Patients with metabolic pH abnormalities should “normally” be expected to develop respiratory compensation as:
- Is there a primary respiratory disorder?
- If we detect a metabolic pH abnormality, there is a possibility that it represents a secondary compensatory response to a respiratory abnormality:
- Metabolic acidosis could be due to chronic respiratory alkalosis.
- Metabolic alkalosis could be due to chronic respiratory acidosis.
- However, this can generally be diagnosed based on a compatible clinical history, and/or prior laboratory studies.
- If we detect a metabolic pH abnormality, there is a possibility that it represents a secondary compensatory response to a respiratory abnormality:
- Is there adequate respiratory compensation?
- Should arterial blood gas measurements be performed in patients with a decreased plasma bicarbonate level when diagnosing acid-base imbalance?
- French guidelines recommend measuring arterial blood gas in every patient with a decreased plasma bicarbonate level (to eliminate respiratory
alkalosis, confirm the diagnosis of metabolic acidosis, and test for mixed acidosis). However, they cite no high-quality evidence to support this recommendation *.
- French guidelines recommend measuring arterial blood gas in every patient with a decreased plasma bicarbonate level (to eliminate respiratory
🔳Read more 📖
- How to convert a VBG into an ABG (PULMCrit)
- ABG/VBG Questions (PULMCrit)
- The diagnostic worthlessness of (most) ABGs (PULMCrit)
- ABG/VBG unhelpful in DKA (PULMCrit)
◾️Considerations
- HCO3 reading in blood gas?
- Note that on blood gas, the HCO3 is calculated and not measured as opposed to blood chemistry.
- Base excess (BE)?
-
- BE corresponds to the quantity of strong acid (or of strong base in the case of metabolic acidosis) that should be added in vitro to 1 L of plasma to normalize the pH to 7.40, with a PaCO2 of 40 mmHg and a temperature of 37 °C.
- Normal range: -5 to +5
- BE means the magnitude and direction of “purely” metabolic contribution to the acid-base disturbances.
- In a patient with a purely metabolic component (e.g. young patients with bleeding secondary to visceral trauma), BE may be helpful to provide magnitude and direction of metabolic derangement, however, in patients with possible respiratory component, BE should not be solely interpreted.
- Measurement of base deficit should probably not be preferred to plasma bicarbonate in identifying patients at risk of metabolic acidosis *.
- ABG vs. VBG?
Blood chemistry
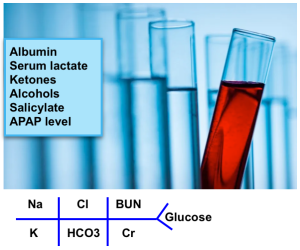
- Fingerstick glucose
- The most notable items are Na+, K+, Cl−, blood urea nitrogen (BUN), creatinine, and albumin.
- Serum lactate *
- Ketones, either urine or serum.
- Salicylate & APAP level.
- CO with oximetry.
- Ethylene glycol & methanol level. Osmolar gap for toxic alcohols.
- Pregnancy test.
Anion Gap (AG)
◾️Background
- Anion gap (AG) is the difference in the concentration between plasma unmeasured anions and cations.
- It quantifies the role of other unmeasured anions in a patient’s acid-base status.
- Always calculate the anion gap (AG) regardless of the pH.
- Almost all acid-base emergencies that require early diagnostic inquiry are in the form of high anion gap acidosis.
- All elevated anion gaps, independent of the patient’s serum bicarbonate or pH, should be carefully examined because many causes of this are immediately life-threatening. (Unlike, for example, non-anion-gap metabolic acidosis, where most causes are not life threats).
◾️Clinical significance: Calculation of AG helps in the following aspects:
- To elucidate causes of metabolic acidosis; high AG or normal AG metabolic acidosis.
- In patients with normal pH, the presence of high AG may be the sole pointer to hidden metabolic acidosis.
- Serial measurement of AG helps in following the effectiveness of treatment, especially in diabetic ketoacidosis.
- Elevated AG may be the only clue to certain clinical disorders.
- For example, D-lactate is not routinely measured in clinical laboratories. The presence of high AG in a patient presenting with neurological issues and a history of short bowel syndrome may be the only point toward D-lactic acidosis.
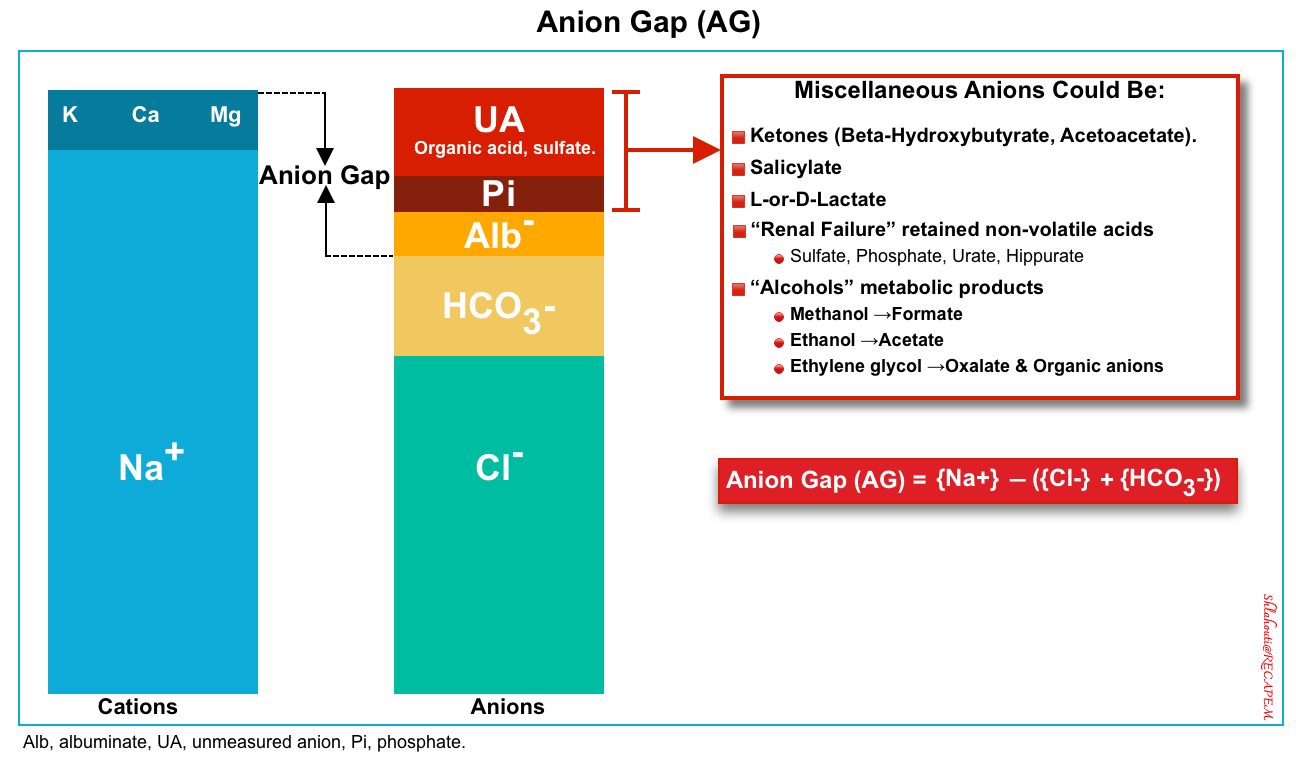
◾️AG calculation
- Every electrolyte panel should have an ion gap evaluated. Ideally, the computer will do this automatically; otherwise, it should be calculated.
- The anion gap is calculated as the following formula. No corrections for (glucose, potassium, etc.).
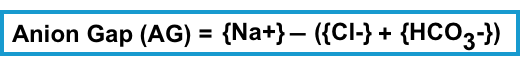
- Is the plasma anion gap corrected for albumin (cAG) better than the uncorrected plasma anion gap in differentiating acid excess from base deficit?
- Albumin is a negatively charged protein, which will theoretically tend to increase the anion gap.
- The physiological AG is mainly composed of phosphate and albuminate (weak anion from blood albumin).
- Every 1g/L decrease in albumin will decrease the anion gap by 0.25 mmoles *.
- Therefore, hypoalbuminemia leads to a decrease in AG.
- In patients with hypoalbuminemia, a normally high anion gap acidosis may appear as a normal anion gap acidosis.
- So, cAG is greater than AG, particularly in a population of patients with a high risk of hypoalbuminemia, as is the case for patients in intensive care or patients with malnutrition, hepatopathy, chronic inflammation, or urinary loss of albumin *.
- If the albumin concentration is reduced, correct the AG as follows:
- Corrected AG = AG + (2.5 x [4.5 – Albumin g/dL]).
- As an example, if the AG is calculated at 15 mEq/L and the albumin concentration is 2.0 g/dL, then the albumin-corrected AG is 21 mEq/L.
- If the albumin concentration is reduced, correct the AG as follows:
- Correcting the anion gap for albumin makes theoretical sense, but it is supported by no clinical evidence *.
- Albumin is a negatively charged protein, which will theoretically tend to increase the anion gap.
◾️AG interpretation
- A normal anion gap is roughly 6-12 meq/L *.
- Normal ranges may vary somewhat between hospital laboratories.
- Historically, the normal range of anion gap was often quoted as being higher (e.g. up to ~16 meq/L). However, with newer electrolyte analyzers, the upper limit of normal has decreased to ~12 meq/L *. For practical purposes, a normal gap can be considered 10 meq/L *.
- Comparison with a baseline anion gap is helpful if that is available. For example:
- Patients with chronic renal insufficiency may often have a chronically elevated anion gap. If their anion gap is at its chronic level, this is reassuring.
- If the anion gap is rapidly elevating over time, this is worrisome and requires aggressive investigation.
- Note that correcting for albumin shifts the anion gap upwards by ~4 mM. Thus, a higher cutoff value must be needed for the detection of an anion-gap metabolic acidosis if correction is made for albumin.
| Causes of high AG |
The serum AG is elevated in those metabolic acidosis due to the accumulation of any strong acid other than hydrochloric acid. The degree to which the AG rises in relation to the fall in bicarbonate varies with the cause of the metabolic acidosis.
🟥Ketoacidosis
- Diabetic ketoacidosis (DKA).
- Alcoholic ketoacidosis (AKA).
- Starvation ketoacidosis
🟥Uremic acidosis
- Occurs when GFR <20-30 ml/min.
- Uncomplicated uremia rarely causes bicarbonate to fall below ~12-15 mM or anion gap to increase over >20 mM (if these are found, look for an alternative or additional disease process).
🟥Hyperlactatemia (see below).
🟥Medication, substance-related
- Toxic alcohols (methanol, ethylene glycol, diethylene glycol, propylene glycol).
- Salicylates.
- Iron.
- Isoniazid.
- Inhalants (i.e., carbon monoxide, cyanide, toluene).
- Sympathomimetics.
- High-dose penicillins.
- Colchicine.
- 5-oxoproline aka pyroglutamic acidosis (therapeutic acetaminophen use) *.
- Usually occurs in women who are ill and malnourished, causing depletion of glutathione and cysteine. Discontinuation of acetaminophen usually leads to rapid resolution *.
🟥Other causes *
- Hyperphosphatemia.
- Marked hyperalbuminemia.
- Metabolic alkalosis (increases the negative charge on albumin).
- D-hyperlactatemia (Due to small intestinal resection or malabsorption that causes carbohydrates to be delivered to the colon and fermented by gut bacteria.
- Note that laboratory tests for “lactate” will not detect D-lactate. D-hyperlactatemia may cause neurological symptoms including confusion, ataxia, incontinence, and nystagmus.
- Anionic paraprotein (usually an immunoglobulin A [IgA] monoclonal immunoglobulin) *.
- Pseudohypobicarbonatemia (an artifactually ↓serum bicarbonate)
- This can occur in the setting of hypertriglyceridemia when bicarbonate is measured spectrophotometrically (likely because triglycerides absorb light). This artifact does not occur when bicarbonate is measured using a blood gas analyzer *.
| Causes of Low AG i.e. AG ≦ 3mM * |
🟥Lab error (likely the most common cause). May occur in
- Severe hypernatremia.
- Pseudohyponatremia (e.g. 2/2 hyperlipidemia).
- Drawing upstream of an IV infusion.
🟥Low albumin level
🟥Increased level of cations
- Hyperkalemia.
- Hypercalcemia.
- Hypermagnesemia.
🟥Elevated immunoglobin level
🟥Pseudohyperchloremia (falsely elevated chloride level) may occur due to:
- Salicylates. (Occurs with central laboratory autoanalyzer utilizing chloride ion selective electrode technology) *.
- Bromide (component of pyridostigmine) *.
- Iodide *.
- Thiosulfate (used in the treatment of calciphylaxis) *.
🟥Other drugs:
Delta-Delta Ratio
◾️Background
- HAGMA may overlap with NAGMA or metabolic alkalosis.
- HCO3– decreases ~1 point for every point increase in AG.
- Various methods exist to assess the additional metabolic component:
- ∆ Anion gap / ∆ HCO3 Ratio, whereas:
- ∆ Anion gap= Calculated AG – 10.
- ∆ HCO3 = 24 – bicarbonate
- Sodium chloride effect (SID), is discussed below.
- ∆ Anion gap / ∆ HCO3 Ratio, whereas:
◾️Clinical significance of “Delta Delta”
- The likely cause of a high AG metabolic acidosis is often apparent from the history, examination, and other laboratory tests (e.g. lactic acidosis in patients with shock, ketoacidosis in patients with hyperglycemia, and uncontrolled type 1 diabetes).
- However, the “Delta Delta” is useful for identifying mixed metabolic disorders and can provide additional information in such patients.
- In patients with high AG metabolic acidosis (HAGMA), the magnitude of the rise in AG relative to the magnitude of the fall in bicarbonate can give some insight into the cause of the metabolic acidosis.
- In pure HAGMA, the change in anion gap (ΔAG) is usually proportional to the change in HCO3 (ΔHCO3). For example, if the anion gap increases by 5 units, the HCO3 will reduce by 5 units. Unequal results signify the presence of mixed metabolic disorders, e.g. metabolic alkalosis plus HAGMA *.
◾️Interpretation
| △AG / △bicarb < 1 |
- High anion gap metabolic acidosis (HAGMA) + normal anion gap metabolic acidosis (NAGMAC), or respiratory alkalosis.
- A ratio of <1 can also be seen in HAGMA in which renal function is preserved and the acid anion is readily excreted into the urine (e.g. ketoacidosis, toluene ingestion, or D-lactic acidosis).
| △AG to △bicarb ≤ 1.6 |
- Ketoacidosis→ Delta Delta ratio =1
- Lactic acidosis→ Delta Delta ratio=1.6
| △AG to △bicarb >1.6 |
- Possible causes are:
Strong Ion Difference (SID)
🔳SID quantifies the role of chloride in a patient’s acid-base status.
🔳SID ~ (Na-Cl). Normal values 38 +/- 2 meq/L.
| SID >40 |
- Vomiting: Loss of HCl (free Cl−).
- Dehydration: Reabsorption of NaHCO3 (free Na+) at the kidney.
- Sodium bicarbonate administration: Increases Na+ in relation to Cl−.
| SID <36 |
- Diarrhea (loss of NaHCO3 ;free Na+; in the GI tract).
- Loss of NaHCO3 in the kidneys (e.g. recovery phase of DKA).
- Renal tubular acidosis.
- Normal saline administration.
- The fluid’s relative concentration of Na+ and Cl− or its SID directly influences the patient’s baseline SID and thus his/her acid-base status *.
- Normal saline has a strong ion difference of 0 (zero). Multiple studies have demonstrated that the administration of normal saline is associated with the development of hyperchloremic (strong ion difference) acidosis.
- Lactated Ringer’s and Plasmalyte have strong ion difference values of 25 and 42, respectively. Such fluids tend to have a neutral or even an alkalizing effect on serum pH.
- High anion gap metabolic acidosis due to accumulation of organic acids with pKa <4 (e.g. lactate, formate, ketoacids).
📖Read more: 9 reasons to quit using normal saline (PULMCrit)
Metabolic Acidosis
General overview
◾️Pathogenesis & Classification
- Metabolic acidosis can be produced by 3 major primary mechanisms:
- ↑Acid generation: Lactic acidosis, ketoacidosis.
- Loss of bicarbonate: Diarrhea or other intestinal losses (eg, tube drainage), proximal (type 2) renal tubular acidosis (RTA), in which proximal bicarbonate reabsorption is impaired.
- ↓Renal acid excretion: Moderate-severe renal insufficiency, Distal (type 1) RTA, and type 4 RTA, in which tubular dysfunction is the primary problem
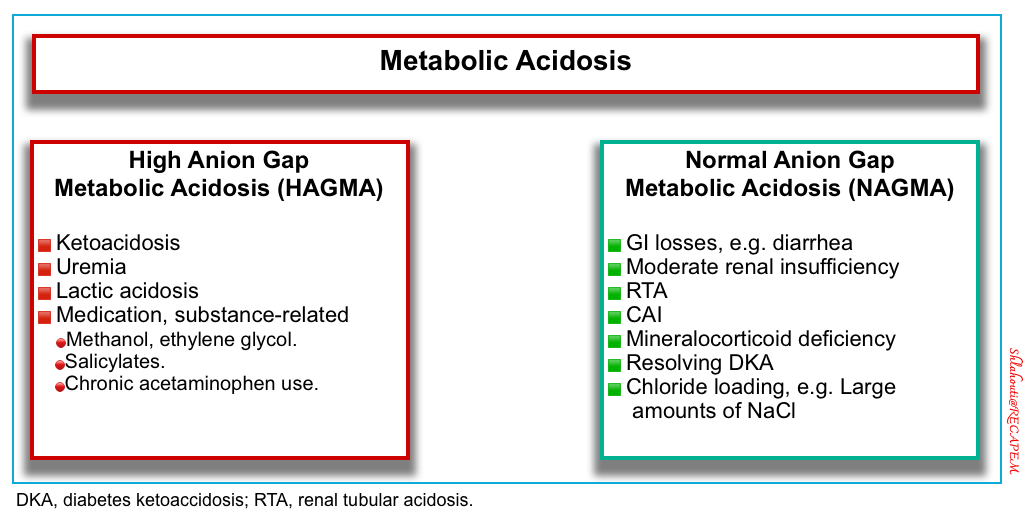
⭐️The serum anion gap can be used to categorize metabolic acidosis into two groups: high anion gap metabolic acidosis and normal anion gap metabolic acidosis
Principles of treatment
◉The treatment of metabolic acidosis varies depending on the underlying disorder. The primary focus of therapy should be directed at reversing the underlying pathophysiologic process (e.g. insulin for DKA).
◉The general principles of metabolic acidosis treatment are covered here, including bicarbonate, hemodialysis, and compensatory hyperventilation for intubated patients.
| 1. Bicarbonate |
Should bicarbonate infusion be used in severe metabolic acidosis and, if so, in what situations?
🔳Background
- Persistent severe acidosis is associated with adverse physiologic impact (discussed above).
- On the other hand, sodium bicarbonate administration is associated with the following risks *
- Hypokalemia (↓K)
- Hypocalcemia (↓iCa)→↓Cardiac contractility, hypotension, seizure, and tetany.
- Hypernatremia (↑Na)
- Rebound alkalemia.
- Water–sodium overload.
- Delay in the fall of blood (lactate: pyruvate) ratio in patients with lactic acidosis.
- ↓O2 delivery to tissues by causing a left shift of the oxygen dissociation curve *.
- Keep in mind that the use of bicarbonate relies on the ability of the patient to have effective ventilation. The bicarbonate anion combines with H ions to ultimately form CO2. If minute ventilation is restricted, preventing adequate exhalation of CO2, excessive administration of bicarbonate may inadvertently cause hypercarbia and paradoxically lower the pH.
| Which metabolic acidosis is most treatable with bicarbonate? |
🔳The utility of bicarbonate administration to patients with severe metabolic acidosis remains controversial, however, this controversy is regarding the use of bicarbonate for the management of lactic acidosis or ketoacidosis *.
-
- In general, giving bicarbonate is recommended for normal anion gap metabolic acidosis (e.g. diarrhea), and uremic high anion gap metabolic acidosis *.
🟥High anion gap metabolic acidosis (HAGMA)
- Shock state and lactic acidosis. Indications of bicarbonate administration after initial resuscitation are:
- Diabetic ketoacidosis
- Sodium bicarbonate should “probably” not be administered to patients with pure diabetic ketoacidosis *. However, giving bicarbonate may be considered in the following clinical situations:
- Uremic high anion gap metabolic acidosis.
- Miscellaneous
- Salicylate poisoning (regardless of pH value) to alkalinize urine. More on this, here 📖.
- Cardiac arrest
- Sodium bicarbonate should not be administered routinely in the therapeutic management of circulatory arrest, apart from hyperkalemic cardiac arrest 📖 or poisoning by membrane stabilizers 📖 *.
- Rhabdomyolysis
- Urinary alkalinization with bicarbonate does not improve patient-centered outcomes in patients with rhabdomyolysis and is not recommended based on the available evidence *. Bicarbonate administration can be considered in those patients with severe non-anion gap metabolic acidosis or uremic acidosis. More on this, here 📖.
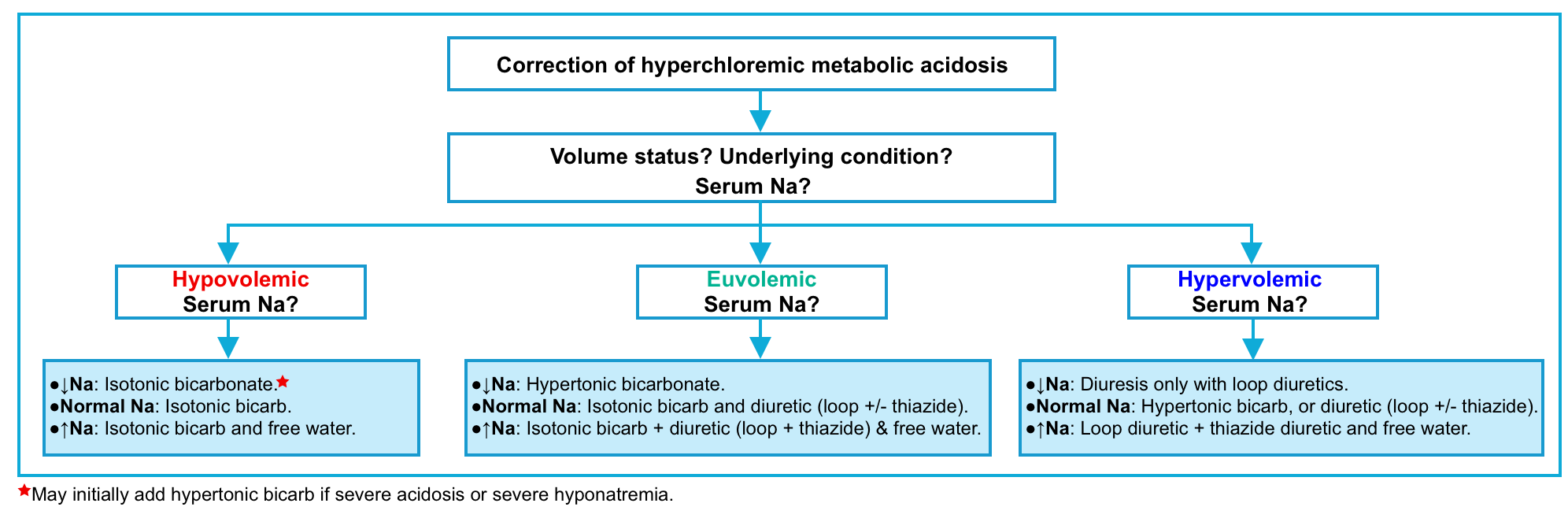
🟩Hyperchloremic metabolic acidosis
- Giving bicarbonate to compensate for gastrointestinal and renal base loss or volume repletion with normal saline is recommended *.
- Hyperchloremia can reduce renal blood flow by causing vasoconstriction of the afferent renal arterioles and altering tubuloglomerular feedback *.
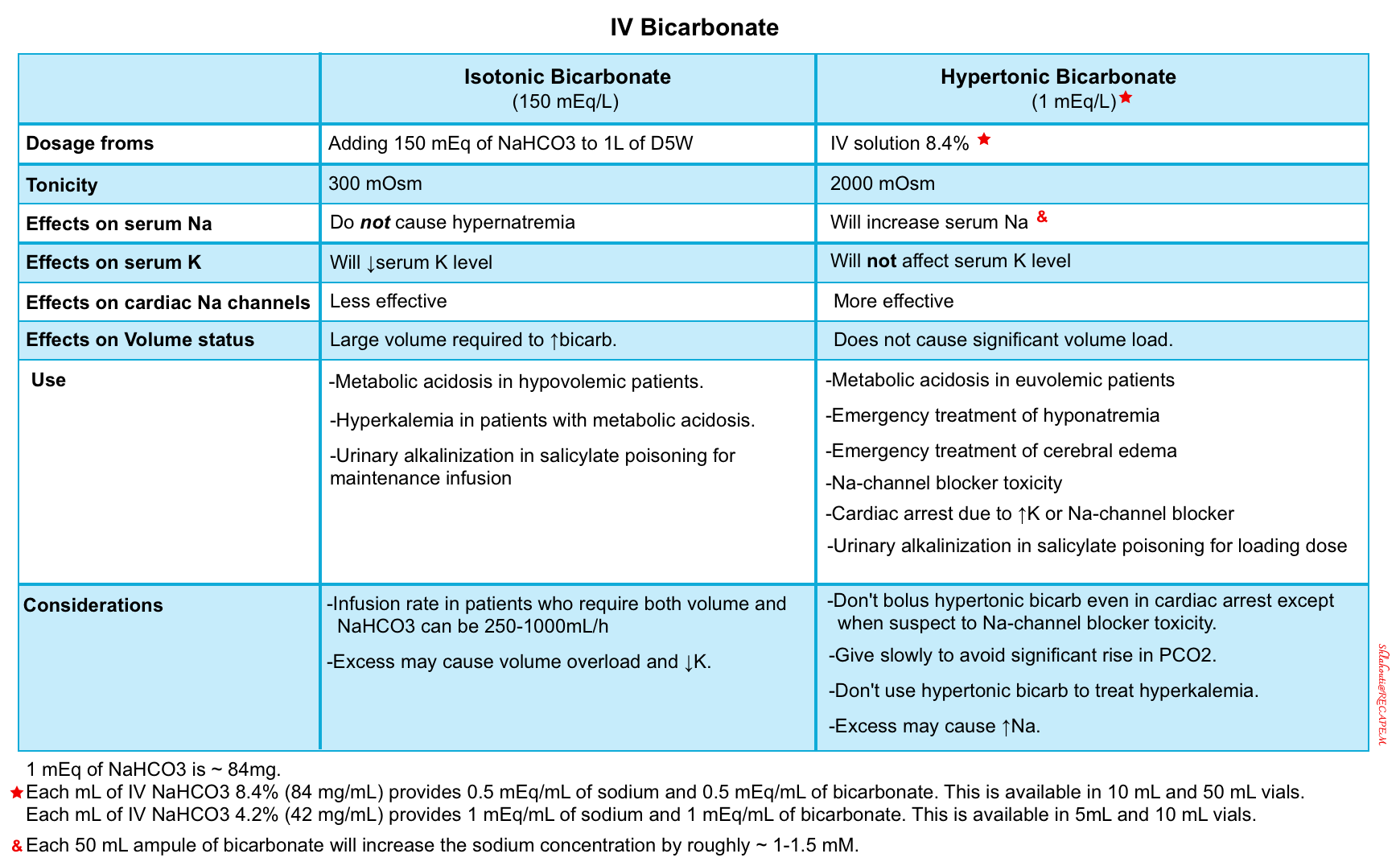
| Principles of bicarbonate administration |
- Avoid hyperchloremic metabolic acidosis. Do not start aggressive IV fluid resuscitation in hypovolemic patients with normal saline.
- Hypertonic bicarbonate: Some or all of this exogenous bicarbonate may be administered as hypertonic sodium bicarbonate (8.4%, described above).
- Hypertonic bicarbonate has the advantage of limiting added volume, but it will eventually cause hypernatremia.
- Isotonic bicarbonate: This may be useful in patients with hypovolemia, hypernatremia, and hyperkalemia.
- NAGMA fundamentally represents an imbalance between sodium chloride and sodium bicarbonate. Treatment therefore may involve the addition of sodium bicarbonate and/or the removal of sodium chloride. The optimal approach depends on the volume status
- Hypovolemia –> Add sodium bicarbonate.
- Euvolemia –> Add sodium bicarbonate and remove sodium chloride simultaneously. The goal here is to replace bicarbonate with chloride, without shifting the volume status.
- Hypervolemia –> Remove sodium chloride via diuresis.
- This gets a bit more complicated because different treatments will affect the sodium concentration (which is fundamentally a reflection of the free water balance). For example:
- Hypertonic bicarbonate will increase the sodium concentration.
- Diuresis using only a loop diuretic (e.g. furosemide) will increase the sodium concentration.
- Diuresis using both a loop diuretic plus a thiazide may facilitate the removal of sodium chloride without causing hypernatremia
| Bicarbonate deficit |
- Bicarbonate deficit = 0.5 x Wt x (Desired 𝚫HCO3–).
- For example, to increase HCO3 from 8 to 15 mEq /L for a 70 kg person
= 0.5 x 70kg x (15-8) = 245 mEq
- For example, to increase HCO3 from 8 to 15 mEq /L for a 70 kg person
- Caution with overshooting if buffering a process that is clearing (e.g. lactate, ketoacidosis, renal failure) →Typically it is safest to only administer until pH is out of a dangerous range (e.g. pH > 7.2, HCO3 > 10-12 mEq/L).
| 2. Hemodialysis (renal replacement therapy, RRT) |
Should RRT be used in severe metabolic acidosis, and if so in what situations?
- Uremic metabolic acidosis
- Severe acidosis (e.g. causing cardiac instability associated with acidosis) refractory to IV bicarbonate, or presence of other indications for RRT 📖 (e.g. refractory hyperkalemia).
- In case of shock and/or acute renal insufficiency, initiation of RRT is suggested if the pH is ≤ 7.15 in the absence of severe respiratory acidosis and despite appropriate treatment *.
- Metabolic acidosis in specific intoxication *
- Metformin poisoning
- In lactic acidosis suggestive of metformin poisoning, early initiation of RRT is suggested when there is an organ dysfunction or in the absence of improvement in the first hours of therapeutic management *.
- Methanol or ethylene glycol poisoning (more on this, here 📖).
- Salicylic acid poisoning (see here).
- Metformin poisoning
| 3. Hyperventilation for intubated patients |
Should minute ventilation be increased in mechanically ventilated patients with metabolic acidosis?
- In metabolic acidosis, the physiological response is an increase in alveolar ventilation leading to a compensatory respiratory alkalosis.
- For intubated patients, the goal is to mimic the normal physiology of compensatory respiratory alkalosis (i.e. targeting a lower pCO2 target than usual).
- Note that the aim of ventilation is not to normalize pH.
- A target pH ≥7.15 seems reasonable *.
- Medical treatment of metabolic acidosis and its cause should be envisaged concomitantly, as ventilatory compensation can only be symptomatic and temporary, and in some patients (e.g. ARDS) not even achievable.
- The experts suggest compensating for acidemia by increasing respiratory frequency without inducing intrinsic PEEP, with a maximum of 35 cycles/min and/or a tidal volume up to 8 mL/kg of body mass, and by monitoring plateau pressure *.
- Note that the extent of hyperventilation will depend on balancing various physiologic derangements:
- In patients with ARDS or obstructive lung disease, achieving a low pCO2 may be impossible or dangerous.
- In patients with healthy lungs and severe metabolic acidosis and hemodynamic instability, there may be a greater inducement to improve the pH by decreasing the pCO2.
High Anion Gap Metabolic Acidosis (HAGMA)
⚠️Elevated anion gap should be regarded as reflecting a life-threatening abnormality until proven otherwise.

1. Ketoacidosis
◾️Definition
- Ketoacidosis is defined as an increase in ketone body concentration, together with a decrease in serum bicarbonate and pH.
- Total ketone levels during normal states are less than 0.5 mmol/L.
- Ketosis is the general term that reflects increased plasma ketones.
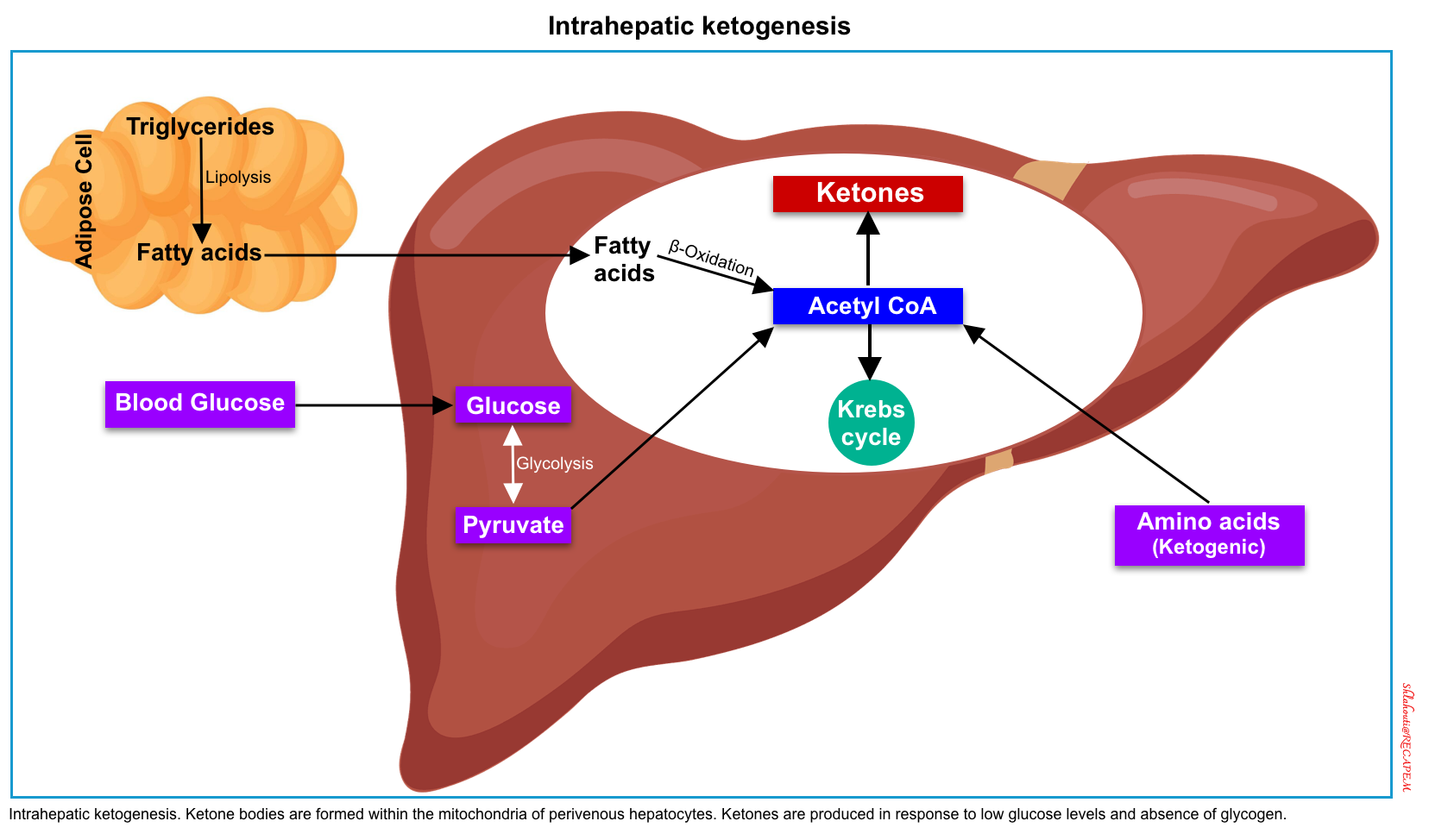
◾️Ketones and ketone metabolism
- Ketogenesis, the production of ketone bodies, takes place in the liver, within the mitochondria of hepatocytes *.
- When carbohydrate availability is significantly reduced, the body resorts to the use of ketones as a primary energy source.
- The 3 ketone bodies which are produced by hepatic β-oxidation of fatty acids (to form acetyl-coenzyme A) are:
- Acetoacetate (AcAc)
- Beta-hydroxybutyrate (BHB)
- Acetone (Ac)
- Of note, acetone is a neutral ketone body and does not cause acidosis or elevated anion gap. In contrast, the two other ketone bodies (acetoacetate and BHB) are acidic and cause ketoacidosis with a high anion gap *.
- The ketone body ratio reflects the amount of BHB relative to AcAc.
- The ratio is almost always > 1.0 and increases markedly with prolonged fasting and with certain pathologic conditions, especially when the intrahepatic redox potential is altered *.
◾️Causes of ketoacidosis
- Metabolic causes
- Toxicogenic causes
- Alcoholic ketoacidosis (AKA)
- Toxic alcohols
- Isopropanol ingestion causes ketosis but does not acidosis.
- Salicylate poisoning
◾️Evaluation
- Blood glucose
- Urinary / blood ketones
- Ketone bodies can be detected by:
- Nitroprusside test reagent which can be applied to urine or serum. However, it’s a qualitative method and can only detect acetoacetate and acetone (BHB is not measurable by NTP)
- Direct measurement of serum BHB
- In the presence of elevated serum glucose level (poor-controlled diabetes), an elevated serum BHB > 3.0 mM/L can be used to diagnose DKA *.
- Ketone bodies can be detected by:
- Blood gas analysis.
- Blood chemistry, anion gap.
- Plasma lactate.
| DKA | Euglycemic DKA | Starvation or High-Protein Diets | AKA | Salicylate Poisoning | Isopropyl Toxicity | |
| Blood glucose | ↑↑↑ | ↑ | ↓ | nl or ↓ | ↓ | – |
| Urinary ketones | ↑↑ | ↑ | ↑ | ↑ | ↑ | ↑↑ |
| Blood Ketones Acetoacetate Acetone BHB | ↑↑ ↑ ↑↑ | ↑↑ ↑ ↑↑ | ↑ ↑ ↑ | ↑↑ ↑ ↑↑↑ | ↑↑ ↑↑ ↑↑ | ↑ ↑↑ ↑ |
| Metabolic Acidosis * | ↑↑ | ↑↑ | – | ↑ | ↑↑ | – |
| ↑Plasma lactate | + | + | + | + | – | – |
| Treatment | here📖 | here📖 | see here 📖 | see here 📖 | Mgmt. of toxic alcohol, 👉here 📖 |
2. Uremia
- The elevated AG metabolic acidosis is predominantly related to several unmeasured strong ions (that are not incorporated into the normal anion gap calculation), including phosphatase, sulfates, and Krebs cycle derivatives.
- High anion gap metabolic acidosis (HAGMAC) occurs when GFR < 20-30 ml/min.
- Uncomplicated uremia rarely causes bicarbonate to fall below ~12-15 mM or anion gap to increase over >20 mM (if these are found, look for an alternative or additional disease process).
- Treatment
- Bicarbonate has been traditionally used to support the pH in efforts to stave off dialysis.
- Evidence has supported the concept of using bicarbonate in uremic metabolic acidosis (to reduce the requirement for dialysis) *.
- Hemodialysis
- This may be indicated in patients with metabolic acidosis and renal failure (especially in the presence of volume overload, which precludes the use of IV bicarbonate).
- According to French guidelines, refractory acidemia (e.g. pH < 7.15 despite conservative measures) might be an indication for dialysis *.
- Bicarbonate has been traditionally used to support the pH in efforts to stave off dialysis.
3. Lactic acidosis
▪️Background
- Lactic acid is an organic acid. It has two enantiomers: L-lactic acid and D-lactic acid.
- L-lactate composes nearly the whole of lactate present in human beings because mammalian cells exclusively contain L-lactate dehydrogenase (LDH); the enzyme that converts pyruvate to lactate (PYR→ Lac).
- In the normal physiologic state, the blood concentration is 0.5-1 mmol/L.
- D-lactate is produced by bacteria. In human beings, this is produced in nanomolar concentrations by GI bacterial flora *. However, it may accumulate in certain pathologic conditions and cause metabolic acidosis. Of note, D-lactic acidosis is not easily identified because D-lactate and L-lactate have the same physical and chemical properties, and the normal analytical methods are not able to distinguish the two isomers *.
- L-lactate composes nearly the whole of lactate present in human beings because mammalian cells exclusively contain L-lactate dehydrogenase (LDH); the enzyme that converts pyruvate to lactate (PYR→ Lac).
- In the following discussion, L-lactate is referred to as lactate unless otherwise specified.
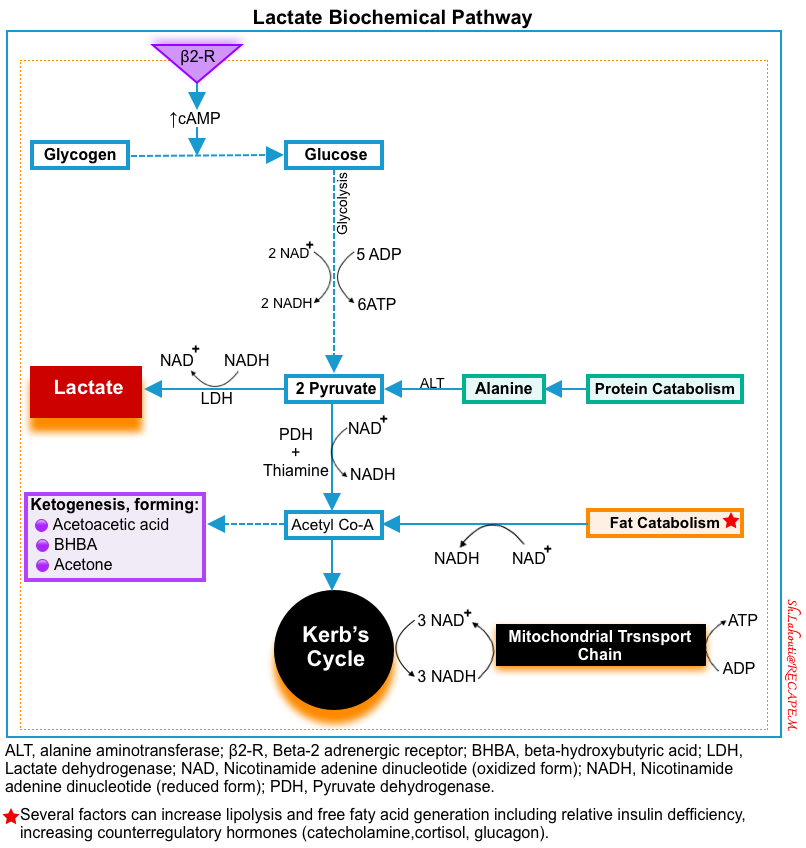
◾️Production of lactic acid
- Source: The main sources of intracellular lactate are glucose (~65%) and alanine (~25%) through their conversion into pyruvate (below figure) *.
- Lactate formation from glucose: In the last step of glycolysis (during normal metabolism), pyruvate is converted to lactate in the cytoplasm by LDH while NADH is oxidized to NAD+ *.
- Lactate formation from alanine: The enzyme alanine aminotransferase (ALT), catalyzes the conversion of alanine to produce pyruvate in the cytoplasm. Pyruvate is then reduced to lactate by LDH *.
- ⭐️This cytosolic regeneration of NAD+ by LDH is mandatory for glycolysis to continue, as NAD+ is needed for the glycolytic reaction that converts glyceraldehyde 3-phosphate into 1,3-bisphosphoglycerate. Without an efficient mechanism in the cytosol to recycle NAD+ from NADH, glycolysis cannot happen *.
◾️Removal of lactic acid
- The lactate is removed principally via two mechanisms *
- Gluconeogenesis (production of glucose from lactate)
- Liver (~60%).
- Renal cortex (~30%)
- Liver (~60%).
- Oxidation as a fuel (10%). This happens in many organs (liver, kidney, muscle, heart, and brain) *.
- Gluconeogenesis (production of glucose from lactate)
▪️Pathogenesis of hyperlactatemia
- Increased lactate production
- Decreased lactate clearance
- Several factors are associated with ↓hepatic clearance, including acidosis, cirrhosis, and hypoperfusion *.
▪️Causes of hyperlactatemia: Hyperlactemia can be divided into two types: Type A (hypoperfusion and tissue hypoxia) vs. Type B (non-hypoxic) *,*. However, the mechanism can be mixed and the same etiology can be found in both groups *.
- Type A: generalized or regional tissue hypoxia *
- Shock of any etiology (septic, cardiogenic, hypovolemic, obstructive).
- Regional hypoperfusion: Mesenteric ischemia, ischemic limbs.
- Muscle hyperactivity, e.g. generalized seizure, extreme exertion.
- Extreme anemia (e.g. <4 g/dL).
- Systemic hypoxemia.
- Type B1: systemic diseases
- Liver failure.
- Thiamine deficiency
- Decompensated diabetes.
- Cancer and hemopathy.
- HIV infection.
- Severe malaria attack *
- Type B2: drugs
- Beta-agonist excess, e.g. epinephrine, albuterol.
- Linezolid.
- Metformin: Virtually, metformin is a mitochondrial toxin, which inhibits the mitochondrial transport chain-I
- Propofol (propofol infusion syndrome).
- Propylene glycol intoxication: Lorazepam, diazepam, nitroglycerine, esmolol, phenytoin, and trimethoprim-sulfamethoxazole.
- Nitroprusside (due to cyanide accumulation).
- Nucleoside reverse-transcriptase inhibitors (e.g., used for the treatment of HCV or HIV) *.
- Oxaliplatin.
- Type B2: toxins
- Alcohols: Ethylene glycol, Methanol, Ethanol poisoning (usually <5 mM).
- Acetaminophen poisoning (massive)
- Carbon monoxide and Cyanide poisoning.
- Salicylate.
- Sympathomimetics (cocaine, amphetamine, cathinone).
- Toluene.
- Iron.
- Type B3: inherited *
- MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes).
- Fructose-1,6-diphosphatase deficiency.
- Glucose-6-phosphatase deficiency (von Gierke disease).
- Pyruvate carboxylase deficiency.
- Pyruvate dehydrogenase deficiency.
- Type D: D-Lactate Excessive production
- Short Bowel Syndrome, jejunoileal bypass surgery.
- In a situation of abdominal compartment syndrome.
- Poisoning from heavy metals.
- Some infections and sepsis *
◾️Evaluation to identify the cause of hyperlactatemia
- Medication list & ingestions
- Physical
- Signs of shock? (tachycardia, low urine output, hypotension, confusion, impaired skin perfusion).
- If there are signs of shock, then resuscitation should begin immediately without delay.
- When in doubt, assume that the hyperlactatemia is real.
- POCUS: Evaluate for unusual types of shock (e.g. massive pulmonary embolism, tamponade).
- Signs of focal ischemia? (abdominal pain or cold limb).
- Signs of shock? (tachycardia, low urine output, hypotension, confusion, impaired skin perfusion).
- Labs
- Anion gap
- ⚠️The anion gap is an insufficient screening tool for elevated lactate levels.
- A significant number (~55%) of ↑lactate levels are not identified when using an anion gap of 12-16 mEq/L *.
- ⚠️The anion gap is an insufficient screening tool for elevated lactate levels.
- Complete blood count
- Elevated white blood cell count or neutrophil/lymphocyte ratio may reflect systemic illness, such as sepsis.
- Severe anemia can increase lactate (but it needs to be extreme).
- Liver function tests (hepatic insufficiency may directly cause or potentiate lactate levels).
- Sepsis evaluation (if sepsis is a consideration):
- Blood cultures.
- Imaging studies (e.g. chest radiographs +/- CT scans).
- Procalcitonin and/or CRP levels.
- Poisoned patient: consider evaluation for:
- Acetaminophen.
- Carbon monoxide.
- Salicylates.
- Toxic alcohols
- Osmolar gap
- It is important to note that the Osm gap is neither a sensitive nor specific test for identifying the presence of a toxic alcohol. A normal osmolar gap does not exclude toxic alcohol poisoning *.
- Anion gap
◾️Treatment
- Treat any identifiable causes of hyperlactatemia.
- Empiric IV thiamine repletion, if deficiency is possible.
- Insulin for insulin-deficient states:
- Insulin increases the activity of PDC (pyruvate dehydrogenase complex), so theoretically, it may decrease lactate levels.
- Insulin administration might be considered for patients with hyperglycemia and borderline ketoacidosis.
- Dialysis or exogenous alkali administration?
- Dialysis is generally ineffective for the management of hyperlactatemia (In vivo, lactic acid is very rapidly being produced and metabolized).
- Dialysis isn’t able to remove lactate rapidly enough to affect this balance. However, dialysis may be considered as a temporizing measure in patients with profound acidemia.
- There is no evidence-based role for bicarbonate in the treatment of elevated lactate levels.
- Dialysis is generally ineffective for the management of hyperlactatemia (In vivo, lactic acid is very rapidly being produced and metabolized).
4. Medication and substance-related
◾️Causes
- Salicylates (more on salicylate acid-base disturbance, here).
- 5-oxoprolinemia/5-oxoprolinuria (pyroglutamic acid) *
-
Mechanism: Glutathione deficiency causes a build-up of gamma-glutamyl cycle metabolites, ie, 5-oxoproline (which is the cause of elevation in anion gap).
-
This condition can be congenital in the pediatric population or may be acquired following acetaminophen use or poisoning; it may be seen in conjunction with certain medications, alcoholism, malnutrition, or critical illness.
-
It often presents as altered mental status or hemolytic anemia.
-
Treatment is stopping the offending agent or treating the underlying clinical condition. N-acetylcysteine can be administered to regenerate glutathione.
-
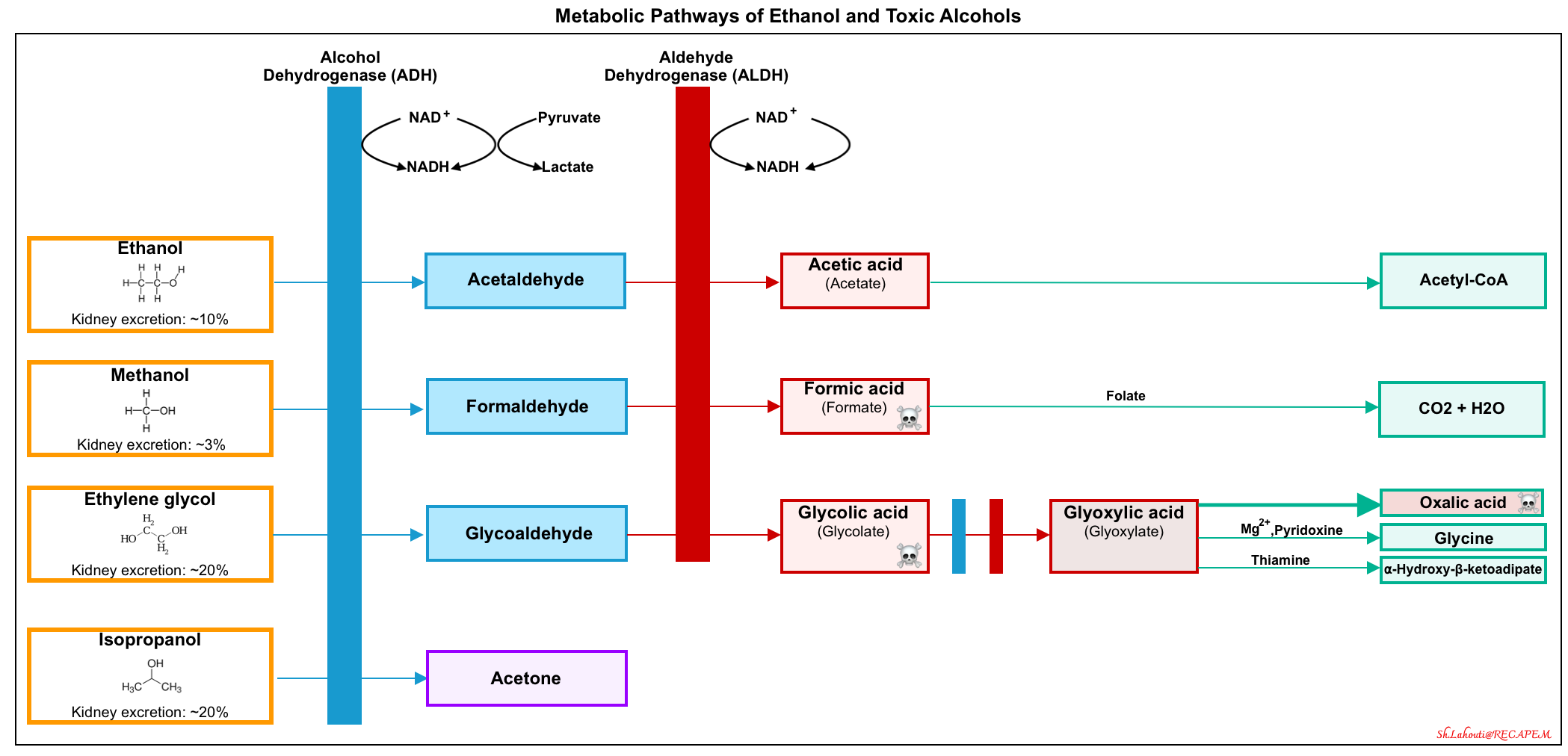
- Methanol and ethylene glycol (for a summary of findings, here). The metabolic pathways of alcohols are shown in the following figure.
Non-Anion Gap Metabolic Acidosis (NAGMA)
◾️Causes
- Gastrointestinal Losses *
- Diarrhea (especially secretory).
- Ureteral diversion.
- Pancreatoenteric fistula or vesiculoenteric fistula.
- Cholestyramine.
- An anion exchange resin that binds bile acids in the gut, can also cause gastrointestinal HCO3− loss and Cl− retention. If the kidneys are unable to compensate, NAGMA develops *.
- Sevelamer hydrochloride.
- An amine anion exchange resin and noncalcium and nonaluminium phosphate binder used widely in patients with advanced CKD is also associated with NAGMA *.
- Kidney
- Moderate renal insufficiency
- Often emerges as GFR falls to ~20-40 ml/min.
- Inadequate ammoniagenesis leads to a NAGMA (if GFR falls further, HAGMA develops).
- Sevelamer use in renal failure may also promote NAGMA.
- Renal tubular acidosis
- Type 1 (distal) RTA (hypokalemic)
- Type 2 (proximal) RTA
- Type 4 RTA (aldosterone resistance or deficiency)
- Carbonic anhydrase inhibitors (acetazolamide and topiramate)*
- Mineralocorticoid deficiency
- Loss of potential bicarbonate, e.g. Resolving diabetic ketoacidosis.
- Urinary loss of organic acid anions, such as ketoacidosis and lactate, can lead to NAGMA. DKA initially presents as HAGMA. As the ketoacidosis resolves, the ketoacids are metabolized to form HCO3−. However, if the ketoacids are lost in the urine before metabolism, this represents the loss of potential HCO3− with the development of NAGMA *.
- Moderate renal insufficiency
- Chloride loading (administration of agents with a low strong ion gap)
- Large amounts of NaCl administration (e.g. normal saline, hypertonic saline) *.
- Large amounts of potassium chloride (either IV or PO).
- Plasma exchange with 4% albumin replacement *.
- Total parenteral nutrition with inadequate acetate.
- In TPN solutions, cationic amino acids lysine and arginine hydrochloride metabolize to HCl, leading to “Cl” load and NAGMA *.
- Calcium chloride administration.
◾️Evaluation
- Complete metabolic profile
- Urinalysis.
- Urine sodium, potassium, glucose, urea (aka “urea nitrogen”), osmolarity, urine pH.
⭐️Identification of mixed HAGMA and NAGMA
- In certain conditions, HAGMA and NAGMA can coexist simultaneously, e.g. patients with recovering DKA, toluene intoxication, CKD, d-lactic acidosis, and profuse diarrhea leading to hypovolemic shock *.
- Calculate Delta Delta ratio
- In simple HAGMA, the Delta Delta ratio is typically ~1.
- A delta ratio of <1 or a delta gap of < 0 signifies the presence of mixed HAGMA and NAGMA *.
| Proximal (type 2) RTA | Distal (type 1) RTA | Hyperkalemic (type 4) RTA | |
| Primary defect | ↓Proximal reabsorption of HCO3 | ↓Distal acid excretion or ↑ H membrane permeability | ↓Excretion of acid and K in the collecting duct |
| Serum HCO3 | 16–20 mmol/L | 10–20 mmol/L | 16–22 mmol/L |
| Serum K | ↓ , e.g. < 3.5 mmol/L | ↓↓, e.g. <<3.5 mmol/L | ↑ (5.5–6.5 mmol/L) |
| Urine pH | <5.5, But will ↑after bicarb load | <5.5, But will ↑after the bicarb load | pH <5.5 with aldosterone deff pH > 5.5 with voltage defect |
| Urine OG | > 150 mOsm | < 150 mOsm | < 150 mOsm |
| Other | Glucosuria Proteinuria may occur. ↓Phos, ↓Ca, ↓Mg, ↓Urate (hypouricemia) | No Glucosuria | No Glucosuria |
| Diagnostic tests | Fractional excretion of HCO3 >15% or urine pH >7.5 after HCO3 loading test | Positive urinary anion gap after NH4 loading test | Urinary K< 40 mmol/L or fractional K excretion <20%, abnormal serum aldosterone, with near- normal renal function. |
| Some causes | Drugs Amphotericin NSAIDs Lithium Foscarnet Metabolic disorder Hyperparathyroidism Hypercalciuria Hyperthyroidism Vit D Intoxication Other Multiple myeloma Nephrotic syndrome Amyloidosis Renal transplant rejection Paroxysmal nocturnal hemoglobinuria Wilson’s disease Sickle cell disease | Drugs Amphotericin NSAIDs Lithium Foscarnet Metabolic disorder Hyperparathyroidism Hypercalciuria Hyperthyroidism Vit D Intoxication Other Toluene abuse Obstructive nephropathy SLE, RA Thyroiditis Multiple myeloma HIV Post-transplant rejection | Drugs TMP, Pentamidine Amiloride, triamterene Spirnolactone, eplerinone ACEI, ARBs, NSAIDs Beta-blockers Cyclosporine, tacrolimus Heparin, Renin inhibitor (aliskiren). Ketoconazole. Metabolic disorder Adrenal insufficiency Other Diabetic nephropathy Hypertensive nephropathy Obstructive nephropathy Multiple myeloma Sickle cell disease HIV Amyloidosis |
| Pharmacotherapy | Alkali therapy (usually K-citrate 10–15 mmol/kg/day), fluids, electrolytes, vitamin D, phosphate, hydrochlorothiazide | NaHCO3 or KHCO3 (1–2 mmol/kg/day), KCl or K-citrate (in patients with severe hypokalemia) | Low-dose fludrocortisone, loop diuretics (if fludrocortisone not tolerated), oral NaHCO3 if serum HCO3 < 22mmol/L, K binders (patiromer or SZC) |
Metabolic Alkalosis
◾️Definition
- Metabolic alkalosis is defined as an increased arterial pH >7.42 or an increase in serum HCO3 to > 30 mmol/L.
◾️Presentation
- Metabolic alkalosis is a common disorder among patients presenting to the emergency department and is generally an asymptomatic laboratory finding.
- Asymptomatic
- Patients with mild to moderate metabolic alkalosis (e.g. serum bicarbonate levels < 40 mmol/L) are typically asymptomatic *.
- When symptoms do occur, they are usually a consequence of an electrolyte abnormality rather than the alkalosis itself; for example:
- Hypokalemia increases the risk of developing cardiac arrhythmias, especially in patients with ischemic heart disease.
- Hypocalcemia (elevated pH shifts calcium ions onto albumin, thereby reducing ionized calcium levels) can cause paresthesias, muscular cramping, and tetany *.
- Neurologic
- Hypoventilation
- As bicarbonate levels increase to 45 mmol/L, the physiologic compensation is to correct the alkalosis by hypoventilation, leading to hypoxemia, especially in patients with a weak respiratory drive (e.g. obesity hypoventilation syndrome or COPD) *.
◾️Pathophysiology
- Mechanism of generation of metabolic alkalosis *
- NaHCO3 or NaHCO3 precursors (i.e., Na acetate, Na citrate, Na gluconate) ingestion, or infusion.
- Distal renal tubule HCO3 generation through enhanced H+ secretion.
- Generous delivery of NaCl (or other Na salts such as Na2SO4 or Na-penicillin) to distal tubules/collecting ducts, which are actively reabsorbing Na+.
- K+ depletion (shifting H+ into cells).
- Removal of HCl from the body (vomiting/nasogastric suction/chloride-rich diarrhea).
- Mechanisms of maintenance *
- Increased proximal renal tubule HCO3 reclamation.
- Hypovolemia.
- ↓K+
- Continuous generation of new HCO3
- In distal kidney tubules and collecting ducts.
- Gastric HCl losses.
- Exogenous alkali.
- Reduced HCO3 filtration: ↓ GFR/kidney failure.
- Increased proximal renal tubule HCO3 reclamation.
◾️Causes
- There are several possible causes of primary metabolic alkalosis. The most common causes are listed in the following table.
- Note that compensatory metabolic alkalosis can occur due to respiratory acidosis, most commonly seen in severe COPD, OHS (obesity hypoventilation syndrome), and chronic respiratory muscle weakness.

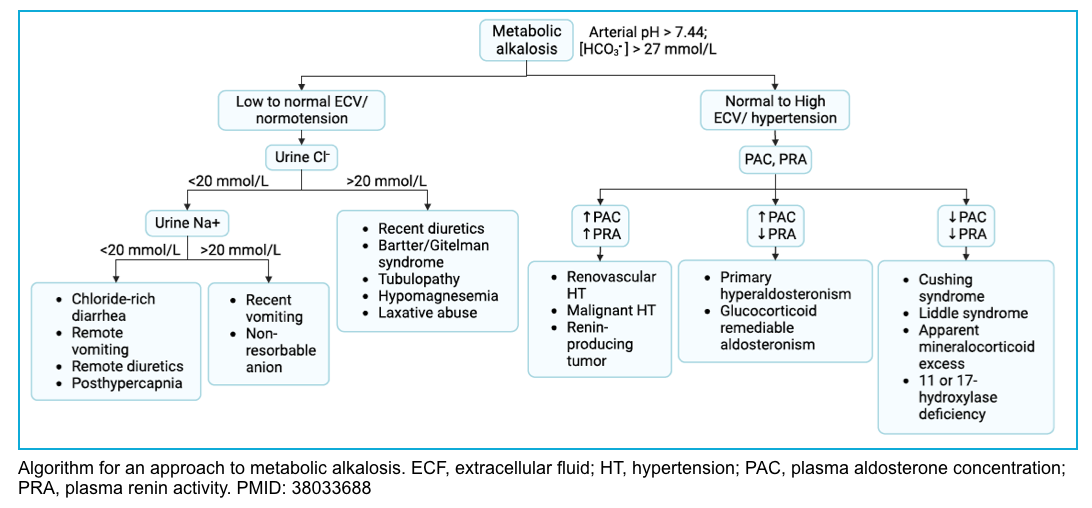
Evaluation and diagnosis
◾️History and physical exams
- A review of available information will usually reveal the cause of the metabolic alkalosis (e.g. a history of vomiting, nasogastric suction, medications, and diuretic use).
- The major decision point in making the diagnosis is based on volume status and blood pressure.
- Hypovolemia (e.g. on bedside echocardiography) suggests chloride deficiency.
- Hypertension suggests excess aldosterone activity.
- Chronicity may be helpful (e.g. a chronic metabolic alkalosis may suggest chronic compensation for COPD).
◾️Evaluation
- If the cause remains unclear further investigation is warranted, including:
- Complete electrolytes (including Ca/Mg/Phos)
- Hypercalcemia may suggest milk-alkali syndrome.
- Hypokalemia and hypomagnesemia may cause metabolic alkalosis (so these require aggressive management).
- Hypophosphatemia might suggest refeeding syndrome as an etiology
- VBG/ABG to evaluate for compensatory metabolic alkalosis.
- If the pCO2 is severely elevated (e.g. >>50 mmHg), this may suggest that there is a primary respiratory acidosis with secondary metabolic compensation.
- Expected respiratory compensation for a primary metabolic alkalosis involves a mild degree of hypercapnia as shown above (e.g. pCO2 ~ 40-50 mmHg). However, this response is highly variable. Hypokalemia may cause diaphragmatic weakness which further elevates the pCO2
- Urine potassium and chloride levels
- Urine potassium <20-30 mM suggests that hypokalemia may be contributory.
- Urine chloride concentration is the most important:
- Chloride <10-30 mM suggests saline responsive.
- Chloride >10-30 mM suggests saline is unresponsive.
- Chloride between 10-30 mM lies in a grey area that doesn’t provide reliable diagnostic information.
- Evaluating Renin-angiotensin-aldosterone system (RAAS)
- Generally, this isn’t useful in critical care. However, this may be considered if:
- Patient has a persistent alkalosis with urine chloride >10-30 mM, and/or is refractory to normal saline infusion.
- Other clinical features suggest excessive activity of the renin-angiotensin-aldosterone system (e.g., hypertension, hypokalemia).
- Investigation of the RAAS involves the measurement of renin & aldosterone levels.
- These may be interpreted as follows *
- Low renin & high aldosterone: Primary hyperaldosteronism (aldosterone-secreting adenoma, bilateral adrenal hyperplasia).
- High renin & high aldosterone: Secondary hyperaldosteronism (renin-secreting tumor, renal artery stenosis).
- Low renin & low aldosterone: State of apparent mineralocorticoid excess (Cushing syndrome, exogenous mineralocorticoid, licorice ingestion).
- Generally, this isn’t useful in critical care. However, this may be considered if:
- Complete electrolytes (including Ca/Mg/Phos)
Treatment
Principles
◾️Identify and treat the underlying cause (s).
- Specific treatment to reduce the bicarbonate is not required in most cases of metabolic alkalosis. Rather, resolving the underlying cause is generally sufficient. For example, a patient with hypovolemia may be treated with volume resuscitation.
- In the following situations, specific treatment of the alkalosis may be indicated:
- Alkalosis is moderate to severe (either causing symptoms).
- The process causing the alkalosis can’t be easily reversed (e.g. patients develop contraction alkalosis from diuretics, but you need to continue diuretic therapy to achieve volume control).
◾️Compensatory metabolic alkalosis due to chronic respiratory acidosis
- This is a compensatory mechanism that is generally beneficial and should not be treated.
- However, if a chronic compensatory alkalosis is exacerbated (e.g. by diuresis), then it may be reasonable to attempt to return the patient to their chronic baseline bicarbonate level.
Multimodal Treatment
A multimodal strategy for severe metabolic alkalosis may be most effective, with attention to all factors perpetuating metabolic alkalosis.

- Discontinue all exogenous sources of alkali to the extent possible.
- This includes sodium or potassium bicarbonate and sodium or potassium salts of any organic anion that is metabolized and thereby generates bicarbonate, such as citrate or lactate.
- Electrolyte repletion if patients have hypokalemia and/or hypomagnesemia
- Potassium chloride should be supplemented aggressively to target a potassium >4.5 mM (unless the patient has renal failure, which places them at increased risk of hyperkalemia).
- Don’t use other types of potassium salts (e.g. potassium citrate or potassium acetate), as the citrate or acetate anions may worsen the alkalosis.
- Correct hypomagnesemia.
- Potassium chloride should be supplemented aggressively to target a potassium >4.5 mM (unless the patient has renal failure, which places them at increased risk of hyperkalemia).
- Start normal saline, if hypovolemic.
- Resuscitation with normal saline may be helpful among patients with hypovolemia (“saline-responsive alkalosis”).
- This is one situation where normal saline is superior to a balanced crystalloid (because you’re looking for an acidotic fluid).
- Resuscitation with normal saline may be helpful among patients with hypovolemia (“saline-responsive alkalosis”).
- Start a diuretic (that prompts bicarbonate excretion), if hypervolemic.
- IV acetazolamide.
- Amiloride.
- Spironolactone. *The main drawback is that it takes ~24-48 hours to work.
- Hold or reduce the dose of diuretic-induced alkalosis (e.g. furosemide).
- If the patient is close to euvolemic, then simply discontinuing alkalosis-inducing diuretics makes sense.
- In patients with mild alkalosis plus persistent volume overload, diuresis should be continued in combination with treatments for alkalosis.
- Proton pump inhibitor in patients with persistent vomiting or NG tube suction *
- Loss of acidic gastric contents will cause metabolic alkalosis.
- Administration of a proton pump inhibitor neutralizes the pH of gastric secretions, preventing the loss of acid via the stomach.
- For patients on TPN, adjust the formulation to remove any sodium acetate.
- Adjust ventilator setting in intubated patients, to target a mild alkalemic pH (e.g. 7.45 -7.50).
- Hypercapnia (a compensatory response) will tend to impair the correction of metabolic alkalosis. Specifically, if the pCO2 is high enough to produce a normal or acidemic pH, this will impair renal bicarbonate excretion.
- To facilitate renal bicarbonate excretion, consider targeting a mildly alkalotic pH (e.g. 7.45-7.50).
- Dialysis
- Alkalosis alone is an exceedingly uncommon indication for dialysis among patients with renal failure. However, dialysis could be reasonable in a patient with other indications for dialysis.
- In a patient with numerous profound electrolyte abnormalities (electrolytic disarray), dialysis will fix all problems simultaneously.
- Alkalosis alone is an exceedingly uncommon indication for dialysis among patients with renal failure. However, dialysis could be reasonable in a patient with other indications for dialysis.
- IV Hydrochloric acid, if all above fail.
- Indications
- Severe metabolic alkalosis (pH >7.55 or bicarbonate >38 meq/L), plus one of the following *:
- Failure of more conservative modalities.
- Alkalosis is so profound that immediate control is needed (insufficient time to use more conservative treatments). Clinical manifestations such as delirium, seizure, or arrhythmia may support a need for immediate therapy.
- The patient remains volume-overloaded, requiring ongoing therapy with diuretics. In this context, HCl may allow for ongoing diuresis with simultaneous management of acid-base status.
- Severe metabolic alkalosis (pH >7.55 or bicarbonate >38 meq/L), plus one of the following *:
- Access
- Must only infused via a central line, ideally via the distal port of the line (if the line gets pulled back a bit, the distal port will remain intravascular) *.
- Confirm the location of the catheter radiographically before the start of the infusion.
- Administration
- Hydrochloric acid is supplied as a 0.1-0.2 Normal solution of HCl (0.1-0.2 mEq/ml). Ideally, this should be formulated in sterile water, to avoid volume overload.
- 0.1 Normal = 100 mEq/L
- 0.2 Normal = 200 mEq/L
- The average safe infusion rate is ~10 mEq/hour *.
- For 0.1 Normal, this is equal to 100 ml/hour.
- For 0.2 Normal, this is equal to 50 ml/hour.
- Monitor electrolytes (including Ca/Mg/Phos) and ABG/VBG (e.g., after every ~75 mEq administered).
- Therapeutic target
- Shoot for a desired bicarbonate above normal (e.g. ~35 mEq/L).
- The goal isn’t to normalize the bicarbonate, but rather merely to remove the patient from immediate danger due to alkalemia.
- Calculate bicarbonate excess:
- Formula for bicarb excess = (0.5)(lean body weight) (plasma bicarb – desired bicarbonate)
- If we’re shooting for a bicarbonate level of ~35 mEq/L, then…
- Bicarb excess = (0.5)(lean body weight)(plasma bicarbonate – 35 mEq/L)
- In a recently published series, the mean amount of HCl infused was 300 mEq.
- Hydrochloric acid is supplied as a 0.1-0.2 Normal solution of HCl (0.1-0.2 mEq/ml). Ideally, this should be formulated in sterile water, to avoid volume overload.
- Indications
Respiratory Acid-base Disorders
-
Traditional methods and caveats
-
In the traditional acid-base teachings, there is a focus on using the blood gas to determine:
-
Whether the respiratory dysfunction (hyper-or hypocarbia) is a primary disorder or a compensatory mechanism.
-
This approach is complex and unreliable.
-
- Whether there is appropriate respiratory compensation for metabolic acidosis by using Winter’s Formula.
- The disadvantages are discussed above.
- The disadvantages are discussed above.
-
-
- Approach in modern clinical practice
-
Do not focus on blood gas to determine if this is a primary disorder or a compensatory mechanism.
-
If pCO2 is elevated, then respiratory acidosis is present.
-
If pCO2 is low, then respiratory alkalosis is present.
-
Use the clinical context to determine whether this imbalance of CO2 production vs. elimination is a primary or secondary problem.
-
- Do not use Winter’s formula to guide diagnosis and management.
- Decisions regarding intubation or the selection of respiratory support devices should generally made based on clinical assessment (e.g. work of breathing, mental status, etc.) and diagnosis, but not the blood gas values.
-
⭐️Physiologic effects of PCO2
| Cerebral | Respiratory | |
| ↑PCO2 | ▪️Vasodilation of cerebral arteries →↑ICP, headache * | ▪️Global vasoconstriction of the pulmonary vasculature →↑PVR. ▪️Acute hypercapnia →Tachypnea manifesting as dyspnea. ▪️Chronic hypercapnia →Blunting of respiratory drive |
| ↓PCO2 | ▪️Vasoconstriction of cerebral arteries resulting in * 1-↓ICP 2-↓Brain perfusion | ▪️Bronchoconstriction & downgrading of hypoxic pulmonary vasoconstriction→ V/Q mismatch may worsen hypoxia. ▪️Blunting the respiratory drive * |
Respiratory Acidosis (Primary Hypercapnia)
◾️Definition
- Respiratory acidosis is acidemia (pH< 7.35) due to an increase in the CO2 tension CO2 (PaCO2 > 45 mm Hg) in bodily fluids.
- Primary hypercapnia is a hypercapnia that isn’t a compensatory response to metabolic alkalosis.
◾️Causes
- “Won’t breathe”: Respiratory mechanics are intact but the patient has lost his/her central drive to maintain CO2 hemostasis *
- Respiratory suppressive medications
- Opioids, benzodiazepines, barbiturates.
- Intoxicants (e.g. ethanol).
- Brainstem dysfunction
- Trauma, CNS infection, tumor.
- Hypothyroidism.
- Central sleep apnea
- Respiratory suppressive medications
- “Can’t breathe”: Central respiratory drive is intact but the patient’s work of breathing has outpaced the patient’s ability to maintain CO2 hemostasis (neuromuscular, chest wall, airway problems) *.
- Spinal cord problem
- C-spine injury (C3-C5), transverse myelitis (e.g. multiple sclerosis), tumor.
- Neuromuscular weakness
- Neuropathy, e.g. Guillain-Barre syndrome, amyotrophic lateral sclerosis, phrenic nerve injury.
- Neuromuscular junction disorder, e.g. Myasthenia gravis, botulism, tick paralysis.
- Myopathy, e.g. hypo-and-hyperkalemia (↓↑K), hypophosphatemia (↓PO4–), thyroid disease, polymyositis, dermatomyositis, muscular dystrophy *.
- Restriction of lung inflation
- Abdominal distension, e.g. abdominal compartment syndrome, OHS (obesity hypoventilation syndrome).
- Pleural disease, e.g. pleural effusion, pneumothorax.
- Thoracic cage dysfunction, e.g. kyphoscoliosis, ankylosing spondylitis, pectus excavatum.
- Upper airway problems, e.g. angioedema, epiglottitis, foreign body, tracheal stenosis, tumor; vocal cord paralysis, or vocal cord dysfunction.
- Small airway problems, e.g. COPD, asthma, bronchiolitis obliterans.
- Spinal cord problem
- “Breathing isn’t working” ( ↑dead space)
- Alveolar disease, e.g. pneumonia, severe interstitial lung disease.
- Vascular disease, e.g. severe pulmonary embolism.
- Mechanical ventilation
◾️Clinical manifestation of hypercapnia
- Acuity of hypercapnia.
- The rapidity of the onset of hypercapnia is often related to the patient’s symptomatology.
- Patients with acute hypercapnia who were previously normocapnic become severely tachypneic manifesting with dyspnea.
- In contrast, significant elevation of PCO2 over the baseline in patients with chronic hypercapnia will blunt the respiratory drive perpetuating the PCO2 elevation. The predominant clinical manifestation is neurologic (Hypercapnia encephalopathy, aka. CO2 narcosis).
- The rapidity of the onset of hypercapnia is often related to the patient’s symptomatology.
- The severity of hypercapnia.
- The clinical signs of hypercapnia also vary depending on the absolute value of PaCO2 and its rate of change *.
- Normal individuals do not develop a depressed level of consciousness until the PaCO2 is > ~80 mm Hg.
- In contrast, patients with chronic hypercapnia (e.g. COPD) may not develop symptoms until PaCO2 is > ~100 mm Hg.
- The clinical signs of hypercapnia also vary depending on the absolute value of PaCO2 and its rate of change *.
- Clinical features
- Tachypnea (more common with acute hypercapnia rather than chronic one).
- CO2 Narcosis (hypercapnic encephalopathy)
- Initially: Anxiety, dyspnea, restlessness.
- Later on: Delirium, somnolence, and eventually coma.
- Other features: coarse tremor, multifocal myoclonus, and asterixis.
- Headache: Nocturnal hypoventilation may cause headache upon awakening.
⭐️While mild-moderate hypercapnia drives the respiratory center, critical hypercapnia has a sedative effect on the central nervous system and patients develop lethargy, and coma (CO2 narcosis).
☠️ In patients with hypoxic respiratory distress, the development of hypercapnia with respiratory acidosis is an ominous sign implying impending respiratory arrest.
◾️Diagnosis
- Diagnosis is based on ABG/VBG.
- Hypercapnia is defined as PaCO2 >45 mmHg.
- Does VBG/ABG provide useful information to differentiate acute vs. chronic vs. acute-on chronic hypercapnia?
- In general, chronic hypercapnia is accompanied by some degree of metabolic alkalosis. However, the traditional teaching of compensatory response does not perfectly answer this question in clinical practice. See the following examples.
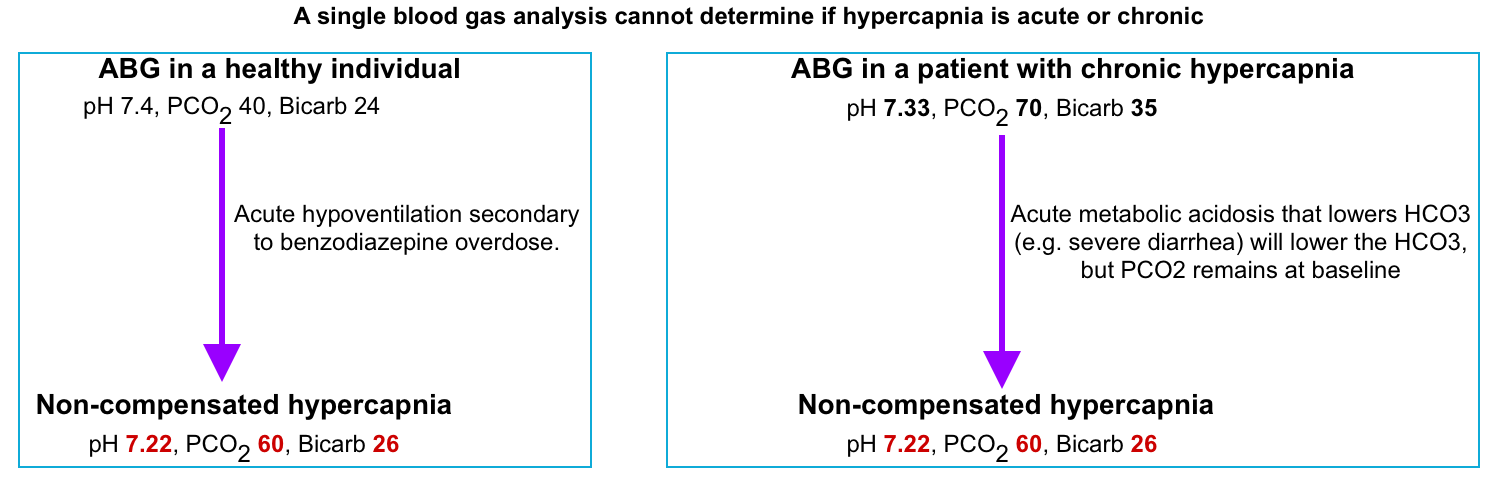
⭐️In clinical practice, the presence of kidney injury which can alter the compensatory mechanism, or the presence of coexisting acid-base disorder often precludes determining the acuity of hypercapnia solely based on compensatory bicarbonate elevation.
◾️Evaluation
- Review comorbidities, medication lists, and archival labs.
- Respiratory rate?
- A low respiratory rate suggests opioid intoxication or a CNS abnormality.
- Respiratory distress?
- Lack of distress suggests either:
- Chronic or acute-on-chronic hypercapnia, with blunting of the respiratory drive.
- Neurologic disease causing a respiratory drive problem.
- Lack of distress suggests either:
- Auscultate over lungs & trachea: Bronchospasm? Stridor? How much air is the patient moving?
- For an intubated patient:
- Peak pressures may suggest airway obstruction below the level of the endotracheal tube. However, this may fail to detect an upper airway obstruction which is bypassed by the endotracheal tube.
- If the patient is hypercapnic despite normal minute ventilation (e.g. ~7-8 liters/minute), this implies an increase in the dead space. More on this, here.
- An elevated gap between the {ABG/VBG pCO2} versus the ETCO2 further supports an increase in dead space.
- Labs
- CBC (polycythemia suggests chronic hypoxemia, often seen with COPD).
- CMP, TSH, Creatinine kinase (may be elevated in myopathies, or hypothyroidism).
- Imaging
- Chest X-ray.
- MRI of the brain/spine, if neurological examination suggests a lesion in those locations.
- CT scan of the neck & lungs (if evidence of upper airway pathology and/or pulmonary dysfunction). CT angiography of the thorax may be indicated if there is a concern for PE as a cause of hypercapnia.
◾️Management
- Treatment depends on the clinical context. Among patients with chronic hypercapnia, the therapeutic goal is generally to return the patient to their baseline pCO2 level.
- Hypercapnia due to opioid intoxication may involve naloxone (depending on the severity; more on this here).
- Hypercapnia due to neuromuscular weakness may often be improved with noninvasive ventilation (e.g., BiPAP). Additionally, the underlying disorders may require specific management.
- Hypercapnia due to asthma or COPD requires specific treatment pathways for these disorders.
- Mild-moderate hypercapnia in an intubated patient may often be monitored without intervention (permissive hypercapnia).
Respiratory Alkalosis (Primary Hypocapnia)
◾️Definition
- Respiratory alkalosis is alkalemia (pH >7.45) due to a decrease in arterial pressure of PaCO2 <35 mm Hg.
- Primary hypocapnia is a hypocapnia that isn’t a compensatory response to metabolic acidosis.
◾️Causes
- Neurologic
- Panic disorder, anxiety, psychosis.
- Central neurogenic hyperventilation, e.g. meningitis, encephalitis, traumatic brain injury, stroke.
- Respiratory disease
- Hypocapnia can be caused by nearly any pulmonary disease.
- Hypoxemia itself can stimulate the respiratory drive, causing hypocapnia.
- Pulmonary irritation can also drive dyspnea and increases in ventilation, likewise leading to hypocapnia.
- Hypocapnia can be caused by nearly any pulmonary disease.
- Toxicologic/medication
- Miscellaneous
- Pain.
- Sepsis.
- This feature results from stimulation of the medullary respiratory center by endotoxins and other inflammatory mediators.
- Hepatic failure.
- Hyperventilation may occur due to hepatic hydrothorax, hepatic encephalopathy, increased progesterone, and estradiol, as well as due to hepatopulmonary syndrome and portopulmonary hypertension *.
- Myxedema coma.
- Recovery from metabolic acidosis.
- Pregnancy (↑progesterone level accounts for the development of respiratory alkalosis in normal pregnancy).
◾️Clinical manifestation
- Central nervous system
- Pathogenesis
- ↓CO2 triggers cerebral vasoconstriction resulting in two major consequences: ↓intracranial pressure and ↓brain perfusion.
- Generally, the dominant effect is a deleterious reduction in brain perfusion.
- Cerebral hypoperfusion due to hypocapnia may contribute to symptoms of a panic attack (e.g. confusion and dizziness).
- In very rare situations with impending herniation, hypocapnia may have therapeutic benefits.
- Signs/symptoms
- Anxiety.
- Delirium, confusion.
- Seizure.
- Pathogenesis
- Cardiovascular
- Arrhythmia.
- Coronary vasospasm with potential to precipitation angina.
- Pulmonary
- Hypocapnia will suppress the respiratory drive.
- Among intubated patients with ventilator-induced hypocapnia, this may cause patients to stop triggering breaths and simply “ride” the ventilator (generally not a desirable situation, as it may promote atelectasis and muscle atrophy).
- Global hypocapnia throughout the lung causes bronchoconstriction and attenuation of hypoxic pulmonary vasoconstriction.
- The net effect is an impairment in ventilation-perfusion matching, which may exacerbate hypoxemia.
- Hypocapnia will suppress the respiratory drive.
- Metabolic
- Hypocalcemia (alkalemia reduces ionized calcium level).
- Paresthesias of extremities and mouth.
- Cramps, carpopedal spasm, rarely tetany.
- Lactic alkalosis
- Hypocapnia stimulates phosphofructokinase (rate-limiting enzyme glycolysis), leading to increased production of lactate.
- Hypophosphatemia (↓PO4-).
- Hypokalemia (↓K).
- Hypocalcemia (alkalemia reduces ionized calcium level).
◾️Evaluation and possible lab findings
- Blood gas: pH >7.45, PaCO2 <35 mm Hg.
- VBG/ABG.
- End-tidal CO2
- An ETCO2 << 30 mmHg suggests hypocapnia.
- However, this may also be caused by pulmonary dysfunction with an increase in dead space volume.
- Therefore, the correlation between the end-tidal CO2 and an ABG/VBG measurement is needed to confirm the diagnosis of hypocapnia.
- Chemistry
- ↓iCa, ↓K, ↓PO4–
- ↑Anion gap (due to lactate production).
- NAGMA (non anion-gap metabolic acidosis) due to metabolic compensation.
- ↓iCa, ↓K, ↓PO4–
◾️Treatment
- Treat any identifiable causes, especially pain and agitation.
Case vignette
Case 1
A 50-year-old woman with a history of insulin-dependent diabetes mellitus was admitted to the ICU in a semi-comatose state. She had been ill for several days. Her medications included subcutaneous insulin and indapamide for hypertension.
On examination, she was barely rousable to verbal command, afebrile, and dehydrated with an HR of 112/min, BP of 94/60 mmHg, and RR of 32/min.
- Initial laboratory investigations revealed Na 132 mmol/L, K 2.7 mmol/L, Cl 79 mmol/L, HCO3 19 mmol/L, blood glucose 543 mg/dl, albumin 4.5g/L, lactate 0.9 mmol/L and a large number of serum ketones.
- Arterial blood gas analysis revealed pH 7.41, PaCO2 32 mmHg, and PaO2 82 mmHg (in room air).
- Question: How do we interpret her blood gases?
- A pH within the normal range despite abnormal values of PCO2 and HCO3 suggests the presence of mixed disorder.
- Calculated AG is 34 [132 − (79 + 19)].
- History of DM, ketonemia, and elevated AG suggest high AG metabolic acidosis due to diabetic ketoacidosis.
- The compensatory response to primary metabolic acidosis:
- The expected PaCO2 will be ~ 38 mmHg (compensation cheat sheet). Since measured PaCO2 < expected PaCO2, there is an associated “Respiratory Alkalosis”.
- Delta AG is 24 (34-10).
- Delta HCO3 is 5 (24-19).
- A (𝚫AG / 𝚫 HCO3) ratio is 4.8; confirming associated “Metabolic Alkalosis”.
- SID is 53 (132-79), which also supports “Metabolic alkalosis”.
- Final diagnosis: High anion gap metabolic acidosis plus metabolic alkalosis.
Going further
Appendix
General features of volume expanders and alkalizing agents
Expected compensation for acid-base disorders

References
1. PMID: 25887061. Kimmoun A, Novy E, Auchet T, Ducrocq N, Levy B. Hemodynamic consequences of severe lactic acidosis in shock states: from bench to bedside. Crit Care. 2015 Apr 9;19(1):175. doi: 10.1186/s13054-015-0896-7. Erratum in: Crit Care. 2017 Feb 21;21(1):40. doi: 10.1186/s13054-017-1624-2.
2. PMID: 28991826. Masevicius FD, et al. . Relationship of Admission Lactate, Unmeasured Anions, and Chloride to the Outcome of Critically Ill Patients. Crit Care Med. 2017 Dec;45(12):e1233-e1239. doi: 10.1097/CCM.0000000000002730.
3. PMID: 26180779. Rodríguez-Gutiérrez R, et al. Severe Ketoacidosis (pH ≤ 6.9) in Type 2 Diabetes: More Frequent and Less Ominous Than Previously Thought. Biomed Res Int. 2015;2015:134780. doi: 10.1155/2015/134780. Epub 2015 Jun 21.
4. PMID: 28149822. Ganesh K, Sharma RN, Varghese J, Pillai MG. A profile of metabolic acidosis in patients with sepsis in an Intensive Care Unit setting. Int J Crit Illn Inj Sci. 2016 Oct-Dec;6(4):178-181. doi: 10.4103/2229-5151.195417.
5. PMID: 23587368. Kajbaf F, Lalau JD. The prognostic value of blood pH and lactate and metformin concentrations in severe metformin-associated lactic acidosis. BMC Pharmacol Toxicol. 2013 Apr 12;14:22. doi: 10.1186/2050-6511-14-22.
6. PMID: 21171991. Friesecke S, Abel P, Roser M, Felix SB, Runge S. Outcome of severe lactic acidosis associated with metformin accumulation. Crit Care. 2010;14(6):R226. doi: 10.1186/cc9376. Epub 2010 Dec 20.
7. Ghosh, S. (2024). Acid-Base Homeostasis: Stewart Approach at the Bedside. In: Malbrain, M.L., Wong, A., Nasa, P., Ghosh, S. (eds) Rational Use of Intravenous Fluids in Critically Ill Patients. Springer, Cham. https://doi.org/10.1007/978-3-031-42205-8_7
8. PMID: 27140683. Story DA. Stewart Acid-Base: A Simplified Bedside Approach. Anesth Analg. 2016 Aug;123(2):511-5. doi: 10.1213/ANE.0000000000001261.
9. PMID: 35074477. Katopodis P, Pappas EM, Katopodis KP. Acid-base abnormalities and liver dysfunction. Ann Hepatol. 2022 Mar-Apr;27(2):100675. doi: 10.1016/j.aohep.2022.100675. Epub 2022 Jan 21.
10. PMID: 30464445. Raveling T, Bladder G, Vonk JM, Nieuwenhuis JA, Verdonk-Struik FM, Wijkstra PJ, Duiverman ML. Improvement in hypercapnia does not predict survival in COPD patients on chronic noninvasive ventilation. Int J Chron Obstruct Pulmon Dis. 2018 Nov 1;13:3625-3634. doi: 10.2147/COPD.S169951.
11. PMID: 22403272. Kraut JA, Madias NE. Differential diagnosis of nongap metabolic acidosis: value of a systematic approach. Clin J Am Soc Nephrol. 2012 Apr;7(4):671-9. doi: 10.2215/CJN.09450911. Epub 2012 Mar 8.
12. PMID: 37675009. Escudero VJ, Mercadal J, Molina-Andújar A, Piñeiro GJ, Cucchiari D, Jacas A, Carramiñana A, Poch E. New Insights Into Diuretic Use to Treat Congestion in the ICU: Beyond Furosemide. Front Nephrol. 2022 Jul 8;2:879766. doi: 10.3389/fneph.2022.879766.
13. PMID: 35525634. Do C, Vasquez PC, Soleimani M. Metabolic Alkalosis Pathogenesis, Diagnosis, and Treatment: Core Curriculum 2022. Am J Kidney Dis. 2022 Oct;80(4):536-551. doi: 10.1053/j.ajkd.2021.12.016. Epub 2022 May 5.
14. PMID: 27931088. Burke KR, Schumacher CA, Harpe SE. SGLT2 Inhibitors: A Systematic Review of Diabetic Ketoacidosis and Related Risk Factors in the Primary Literature. Pharmacotherapy. 2017 Feb;37(2):187-194. doi: 10.1002/phar.1881. Epub 2017 Jan 16.
15. PMID: 34979145. Giaccari A, Pontremoli R, Perrone Filardi P. SGLT-2 inhibitors for the treatment of heart failure in patients with and without type 2 diabetes: A practical approach for routine clinical practice. Int J Cardiol. 2022 Mar 15;351:66-70. doi: 10.1016/j.ijcard.2021.12.050. Epub 2022 Jan 1.
16. PMID: 31418093. Jung B, Martinez M, Claessens YE, Darmon M, Klouche K, Lautrette A, Levraut J, Maury E, Oberlin M, Terzi N, Viglino D, Yordanov Y, Claret PG, Bigé N; Société de Réanimation de Langue Française (SRLF); Société Française de Médecine d’Urgence (SFMU). Diagnosis and management of metabolic acidosis: guidelines from a French expert panel. Ann Intensive Care. 2019 Aug 15;9(1):92. doi: 10.1186/s13613-019-0563-2.
17. PMID: 35998977. Achanti A, Szerlip HM. Acid-Base Disorders in the Critically Ill Patient. Clin J Am Soc Nephrol. 2023 Jan 1;18(1):102-112. doi: 10.2215/CJN.04500422. Epub 2022 Aug 23.
18. PMID: 23903783. Bloom BM, Grundlingh J, Bestwick JP, Harris T. The role of venous blood gas in the emergency department: a systematic review and meta-analysis. Eur J Emerg Med. 2014 Apr;21(2):81-8. doi: 10.1097/MEJ.0b013e32836437cf.
19. PMID: 30306080. Montagnana M, Danese E, Lippi G. Biochemical markers of acute intestinal ischemia: possibilities and limitations. Ann Transl Med. 2018 Sep;6(17):341. doi: 10.21037/atm.2018.07.22.
20. PMID: 6166764. Nanji AA, Campbell DJ, Pudek MR. Decreased anion gap associated with hypoalbuminemia and polyclonal gammopathy. JAMA. 1981 Aug 21;246(8):859-60.
21. PMID: 18431828. Chawla LS, Jagasia D, et al. Anion gap, anion gap corrected for albumin, and base deficit fail to accurately diagnose clinically significant hyperlactatemia in critically ill patients. J Intensive Care Med. 2008 Mar-Apr;23(2):122-7. doi: 10.1177/0885066607312985.
22. PMID: 16858097. Dinh CH, Ng R, Grandinetti A, Joffe A, Chow DC. Correcting the anion gap for hypoalbuminemia does not improve the detection of hyperlactatemia. Emerg Med J. 2006 Aug;23(8):627-9. doi: 10.1136/emj.2005.031898.
23. PMID: 21277142. Mallat J, Michel D, Salaun P, Thevenin D, Tronchon L. Defining metabolic acidosis in patients with septic shock using Stewart approach. Am J Emerg Med. 2012 Mar;30(3):391-8. doi: 10.1016/j.ajem.2010.11.039. Epub 2011 Jan 28.
24. PMID: 19087326. Chawla LS, Shih S, Davison D, Junker C, Seneff MG. Anion gap, anion gap corrected for albumin, base deficit and unmeasured anions in critically ill patients: implications on the assessment of metabolic acidosis and the diagnosis of hyperlactatemia. BMC Emerg Med. 2008 Dec 16;8:18. doi: 10.1186/1471-227X-8-18.
25. PMID: 24766940. Rice M, Ismail B, Pillow MT. Approach to metabolic acidosis in the emergency department. Emerg Med Clin North Am. 2014 May;32(2):403-20. doi: 10.1016/j.emc.2014.01.002. Epub 2014 Feb 25.
26. PMID: 31565630. Venkataraman SS, Regone R, Ammar HM, Govindu RR. Pyroglutamic Acidemia: An Underrecognized and Underdiagnosed Cause of High Anion Gap Metabolic Acidosis – A Case Report and Review of Literature. Cureus. 2019 Jul 24;11(7):e5229. doi: 10.7759/cureus.5229
27. PMID: 34400023. enves AZ, Emmett M. Approach to Patients With High Anion Gap Metabolic Acidosis: Core Curriculum 2021. Am J Kidney Dis. 2021 Oct;78(4):590-600. doi: 10.1053/j.ajkd.2021.02.341. Epub 2021 Aug 13.
28. PMID: 17699401. Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007 Jan;2(1):162-74. doi: 10.2215/CJN.03020906. Epub 2006 Dec 6.
29. PMID: 22157711. van Hoeven KH, Joseph RE, Gaughan WJ, McBride L, Bilotti E, McNeill A, Schmidt L, Schillen D, Siegel DS. The anion gap and routine serum protein measurements in monoclonal gammopathies. Clin J Am Soc Nephrol. 2011 Dec;6(12):2814-21. doi: 10.2215/CJN.07380711.
30. PMID: 37661152. Akkawi AR, Fawcett M, Challa A, Nguyen T, Jackson J, Muraga R. Hypertriglyceridemia-Induced Pseudo-High Anion Gap Metabolic Acidosis Manifesting as Ischemic Stroke. Mayo Clin Proc. 2023 Sep;98(9):1425-1426. doi: 10.1016/j.mayocp.2023.07.012.
31. PMID: 37783490. Haber LA, Dhaliwal G, Lo L, Rizzuto G. Evaluating a low anion gap: A practical approach. Cleve Clin J Med. 2023 Oct 2;90(10):619-623. doi: 10.3949/ccjm.90a.23035.
32. PMID: 33864761. Wiederkehr MR, Benevides R Jr, Santa Ana CA, Emmett M. Pseudohyperchloremia and Negative Anion Gap – Think Salicylate! Am J Med. 2021 Sep;134(9):1170-1174. doi: 10.1016/j.amjmed.2021.03.017. Epub 2021 Apr 20.
33. PMID: 26363848. Emmett M. Approach to the Patient With a Negative Anion Gap. Am J Kidney Dis. 2016 Jan;67(1):143-50. doi: 10.1053/j.ajkd.2015.07.024. Epub 2015 Sep 10.
34. PMID: 29344509. Berend K. Review of the Diagnostic Evaluation of Normal Anion Gap Metabolic Acidosis. Kidney Dis (Basel). 2017 Dec;3(4):149-159. doi: 10.1159/000479279. Epub 2017 Sep 1.
35. PMID: 28796408. Komaru Y, Inokuchi R, Ueda Y, Nangaku M, Doi K. Use of the anion gap and intermittent hemodialysis following continuous hemodiafiltration in extremely high dose acute-on-chronic lithium poisoning: A case report. Hemodial Int. 2018 Jan;22(1):E15-E18. doi: 10.1111/hdi.12583. Epub 2017 Aug 10.
36. PMID: 36066292. Bajwa GS, Hussain A, Javaid MM. How to work up an adult patient with metabolic acidosis. Br J Hosp Med (Lond). 2022 Aug 2;83(8):1-11. doi: 10.12968/hmed.2021.0582. Epub 2022 Aug 5.
37. PMID: 15774079. Morgan TJ. The meaning of acid-base abnormalities in the intensive care unit: part III — effects of fluid administration. Crit Care. 2005 Apr;9(2):204-11. doi: 10.1186/cc2946. Epub 2004 Sep 3.
38. PMID: 22945490. Kraut JA, Madias NE. Treatment of acute metabolic acidosis: a pathophysiologic approach. Nat Rev Nephrol. 2012 Oct;8(10):589-601. doi: 10.1038/nrneph.2012.186. Epub 2012 Sep 4.
39. PMID: 38616273. Vidanapathirana M. Sodium bicarbonate and intubation in severe diabetic ketoacidosis: are we too quick to dismiss them? Clin Diabetes Endocrinol. 2024 Apr 15;10(1):13. doi: 10.1186/s40842-024-00171-y.
40. PMID: 18322160. Sabatini S, Kurtzman NA. Bicarbonate therapy in severe metabolic acidosis. J Am Soc Nephrol. 2009 Apr;20(4):692-5. doi: 10.1681/ASN.2007121329. Epub 2008 Mar 5.
41. PMID: 29910040. Jaber S, et.al ; BICAR-ICU Study Group. Sodium bicarbonate therapy for patients with severe metabolic acidaemia in the intensive care unit (BICAR-ICU): a multicentre, open-label, randomized controlled, phase 3 trial. Lancet. 2018 Jul 7;392(10141):31-40. doi: 10.1016/S0140-6736(18)31080-8. Epub 2018 Jun 14. Erratum in: Lancet. 2018 Dec 8;392(10163):2440. doi: 10.1016/S0140-6736(18)33040-X.
42. PMID: 37442665. Wardi G, Holgren S, Gupta A, Sobel J, Birch A, Pearce A, Malhotra A, Tainter C. A Review of Bicarbonate Use in Common Clinical Scenarios. J Emerg Med. 2023 Aug;65(2):e71-e80. doi: 10.1016/j.jemermed.2023.04.012. Epub 2023 Apr 21.
43. PMID: 18322160. Sabatini S, Kurtzman NA. Bicarbonate therapy in severe metabolic acidosis. J Am Soc Nephrol. 2009 Apr;20(4):692-5. doi: 10.1681/ASN.2007121329. Epub 2008 Mar 5.
44. PMID: 33851122. Melamed ML, Raphael KL. Metabolic Acidosis in CKD: A Review of Recent Findings. Kidney Med. 2021 Feb 10;3(2):267-277. doi: 10.1016/j.xkme.2020.12.006.
45. PMID: 22998993. Cartwright MM, Hajja W, Al-Khatib S, Hazeghazam M, Sreedhar D, Li RN, Wong-McKinstry E, Carlson RW. Toxigenic and metabolic causes of ketosis and ketoacidosis syndromes. Crit Care Clin. 2012 Oct;28(4):601-31. doi: 10.1016/j.ccc.2012.07.001.
46. PMID: 23131345. Frise CJ, Mackillop L, Joash K, Williamson C. Starvation ketoacidosis in pregnancy. Eur J Obstet Gynecol Reprod Biol. 2013 Mar;167(1):1-7. doi: 10.1016/j.ejogrb.2012.10.005. Epub 2012 Nov 4.
47. PMID: 31019864. Mubarik A, Jupalli A, Iqbal AM, Muddassir S, Eddib A. Isolated Starvation Ketoacidosis: A Rare Cause of Severe Metabolic Acidosis Presenting with a pH Less than 7. Cureus. 2019 Feb 17;11(2):e4086. doi: 10.7759/cureus.4086.
48. PMID: 19564476. Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009 Jul;32(7):1335-43. doi: 10.2337/dc09-9032.
49. PMID: 16507145. Gunnerson KJ, Saul M, He S, Kellum JA. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Crit Care. 2006 Feb;10(1):R22. doi: 10.1186/cc3987.
50. PMID: 31039813. Liu Z, Meng Z, Li Y, Zhao J, Wu S, Gou S, Wu H. Prognostic accuracy of the serum lactate level, the SOFA score and the qSOFA score for mortality among adults with Sepsis. Scand J Trauma Resusc Emerg Med. 2019 Apr 30;27(1):51. doi: 10.1186/s13049-019-0609-3.
51. PMID: 24929216. Adeva-Andany M, López-Ojén M, Funcasta-Calderón R, Ameneiros-Rodríguez E, Donapetry-García C, Vila-Altesor M, Rodríguez-Seijas J. Comprehensive review on lactate metabolism in human health. Mitochondrion. 2014 Jul;17:76-100. doi: 10.1016/j.mito.2014.05.007. Epub 2014 Jun 12.
52. PMID: 31784049. Kamel KS, Oh MS, Halperin ML. L-lactic acidosis: pathophysiology, classification, and causes; emphasis on biochemical and metabolic basis. Kidney Int. 2020 Jan;97(1):75-88. doi: 10.1016/j.kint.2019.08.023. Epub 2019 Sep 6.
53. PMID: 31784049. Kamel KS, Oh MS, Halperin ML. L-lactic acidosis: pathophysiology, classification, and causes; emphasis on biochemical and metabolic basis. Kidney Int. 2020 Jan;97(1):75-88. doi: 10.1016/j.kint.2019.08.023. Epub 2019 Sep 6.
54. PMID: 36558947. Zanza C, Facelli V, Romenskaya T, Bottinelli M, Caputo G, Piccioni A, Franceschi F, Saviano A, Ojetti V, Savioli G, Longhitano Y. Lactic Acidosis Related to Pharmacotherapy and Human Diseases. Pharmaceuticals (Basel). 2022 Nov 30;15(12):1496. doi: 10.3390/ph15121496.
55. PMID: 31474479. Wardi G, Brice J, Correia M, Liu D, Self M, Tainter C. Demystifying Lactate in the Emergency Department. Ann Emerg Med. 2020 Feb;75(2):287-298. doi: 10.1016/j.annemergmed.2019.06.027. Epub 2019 Aug 29. Erratum in: Ann Emerg Med. 2020 Apr;75(4):557. doi: 10.1016/j.annemergmed.2020.02.021.
56. PMID: 26378980. Suetrong B, Walley KR. Lactic Acidosis in Sepsis: It’s Not All Anaerobic: Implications for Diagnosis and Management. Chest. 2016 Jan;149(1):252-61. doi: 10.1378/chest.15-1703. Epub 2016 Jan 6.
57. PMID: 36558947. Zanza C, Facelli V, Romenskaya T, Bottinelli M, Caputo G, Piccioni A, Franceschi F, Saviano A, Ojetti V, Savioli G, Longhitano Y. Lactic Acidosis Related to Pharmacotherapy and Human Diseases. Pharmaceuticals (Basel). 2022 Nov 30;15(12):1496. doi: 10.3390/ph15121496.
58. PMID: 17116255. Jørgensen VL, Reiter N, Perner A. Luminal concentrations of L- and D-lactate in the rectum may relate to the severity of disease and outcome in septic patients. Crit Care. 2006;10(6):R163. doi: 10.1186/cc5102.
59. PMID: 28736054. Xu Q, HowlettClyne S, Fuezery A, Cembrowski GS. Low sensitivity of anion gap to detect clinically significant lactic acidosis in the emergency department. Clin Biochem. 2017 Dec;50(18):1164-1167. doi: 10.1016/j.clinbiochem.2017.07.008. Epub 2017 Jul 20.
60. PMID: 18045860. Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. Clin J Am Soc Nephrol. 2008 Jan;3(1):208-25. doi: 10.2215/CJN.03220807. Epub 2007 Nov 28.
61. PMID: 38837536. Bhandari R, Ekladious A, Javaid MM. Demystifying normal-anion-gap metabolic acidosis: pathophysiology, etiology, evaluation, and diagnosis. Intern Med J. 2024 Jul;54(7):1056-1065. doi: 10.1111/imj.16418. Epub 2024 Jun 5.
62. PMID: 24766943. Soifer JT, Kim HT. Approach to metabolic alkalosis. Emerg Med Clin North Am. 2014 May;32(2):453-63. doi: 10.1016/j.emc.2014.01.005. Epub 2014 Mar 14.
63. PMID: 35525634. Do C, Vasquez PC, Soleimani M. Metabolic Alkalosis Pathogenesis, Diagnosis, and Treatment: Core Curriculum 2022. Am J Kidney Dis. 2022 Oct;80(4):536-551. doi: 10.1053/j.ajkd.2021.12.016. Epub 2022 May 5.
64. PMID: 32586924. Emmett M. Metabolic Alkalosis: A Brief Pathophysiologic Review. Clin J Am Soc Nephrol. 2020 Dec 7;15(12):1848-1856. doi: 10.2215/CJN.16041219. Epub 2020 Jun 25.
65. PMID: 30369299. Gillion V, Jadoul M, Devuyst O, Pochet JM. The patient with metabolic alkalosis. Acta Clin Belg. 2019 Feb;74(1):34-40. doi: 10.1080/17843286.2018.1539373. Epub 2018 Oct 27.
66. PMID: 17140170. Kirsch BM, Sunder-Plassmann G, Schwarz C. Metabolic alkalosis in a hemodialysis patient–successful treatment with a proton pump inhibitor. Clin Nephrol. 2006 Nov;66(5):391-4. doi: 10.5414/cnp66391.
67. PMID: 29359573. Guffey JD, Haas CE, Crowley A, Connor KA, Kaufman DC. Hydrochloric Acid Infusion for the Treatment of Metabolic Alkalosis in Surgical Intensive Care Unit Patients. Ann Pharmacother. 2018 Jun;52(6):522-526. doi: 10.1177/1060028018754389. Epub 2018 Jan 23.
68. PMID: 15693984. O’Croinin D, Ni Chonghaile M, Higgins B, Laffey JG. Bench-to-bedside review: Permissive hypercapnia. Crit Care. 2005 Feb;9(1):51-9. doi: 10.1186/cc2918. Epub 2004 Aug 5.
69. PMID: 26976648. Davidson AC, et al; BTS Standards of Care Committee Member, British Thoracic Society/Intensive Care Society Acute Hypercapnic Respiratory Failure Guideline Development Group, On behalf of the British Thoracic Society Standards of Care Committee. BTS/ICS guideline for the ventilatory management of acute hypercapnic respiratory failure in adults. Thorax. 2016 Apr;71 Suppl 2:ii1-35. doi: 10.1136/thoraxjnl-2015-208209. Erratum in: Thorax. 2017 Jun;72(6):588. doi: 10.1136/thoraxjnl-2015-208209corr1.
70. PMID: 37341662. Palmer BF, Clegg DJ. Respiratory Acidosis and Respiratory Alkalosis: Core Curriculum 2023. Am J Kidney Dis. 2023 Sep;82(3):347-359. doi: 10.1053/j.ajkd.2023.02.004. Epub 2023 Jun 21. Erratum in: Am J Kidney Dis. 2024 Jan;83(1):126. doi: 10.1053/j.ajkd.2023.10.005.
71. PMID: 35074477. Katopodis P, Pappas EM, Katopodis KP. Acid-base abnormalities and liver dysfunction. Ann Hepatol. 2022 Mar-Apr;27(2):100675. doi: 10.1016/j.aohep.2022.100675. Epub 2022 Jan 21.

Add comment