


July 14, 2021, via Shahriar Lahouti
CONTENTS
- Preface
- Clinical significance of bradycardia
- Etiology
- Clinical manifestation
- General approach in ED
- Fundamental concepts
- Evaluation
- Resuscitation
- RECAP
- Going further
- Reference
Preface
Severe progressive sinus bradycardia is a common pathway of impending death from any terminal disease state. In this post, the principles of management of unstable patients with bradycardia are discussed.
Clinical significance of bradycardia
Bradycardia is defined as a heart rate of less than 50 bpm 1 in adults (non-well-conditioned athletes). Generally severe bradycardia is more worrisome than tachycardia for the following reasons:
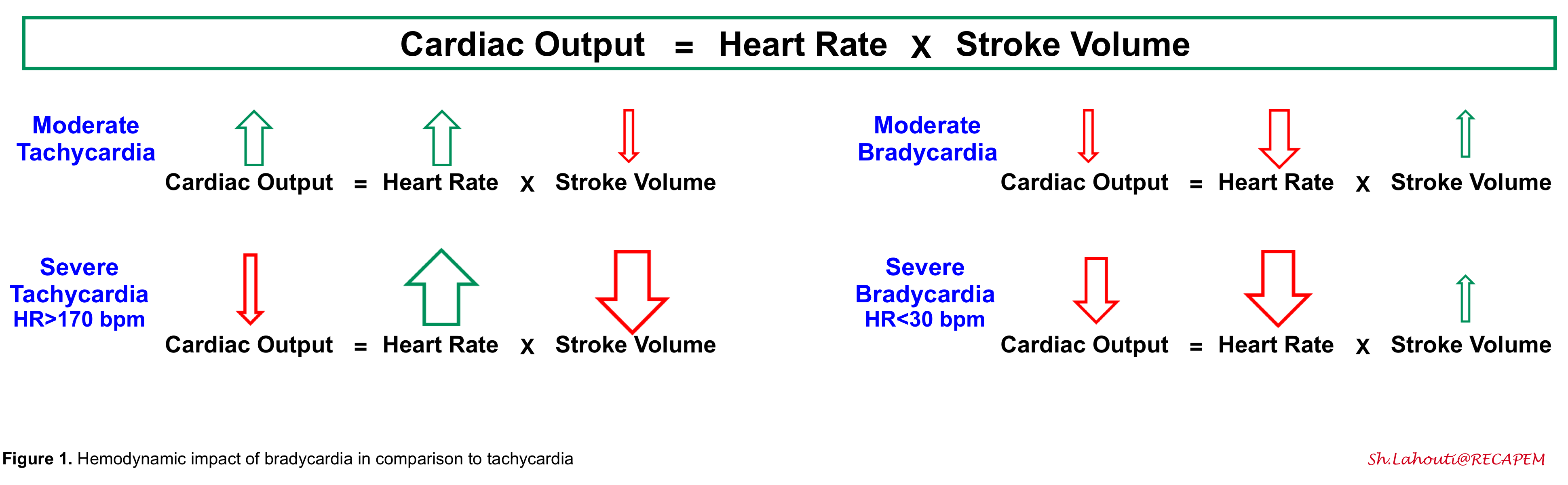
1.Cardiac output is dependent on heart rate and stroke volume. In mild-moderate tachycardia, the effect of the increased heart rate predominates, and cardiac output is increased. However, in severe tachycardia, the cardiac output drops since the diastolic filling time is reduced (causing reduced stroke volume). Bradycardia can directly pull down cardiac output and cause hemodynamic instability 2. Theoretically, slowing down the heart rate may cause a minimal increase in diastolic filling, thereby increasing the stroke volume. However, this compensatory factor is weak and extremely limited.
2.Progressive bradycardia is often a harbinger of death☠️
3.Torsade de pointes is a pause-dependent arrhythmia, which is more likely to occur at slower heart rates. Moreover, bradycardia itself may prolong the QT interval. Leaving patients in a severely bradycardic state may increase their risk of torsade.
Etiology
◾️Medications 3
- Beta-blockers (including eye drops for glaucoma, e.g., timolol), and calcium channel blockers.
- Digoxin.
- Antiarrhythmics (class I and III).
- Central alpha-2 agonist (e.g., clonidine, dexmedetomidine).
- Cholinergic agents.
- Direct receptor agonist: Bethanechol
- Indirect-acting (working at synapses, i.e., acetylcholine esterase inhibitors)
- Edrophonium, neostigmine, pyridostigmine, rivastigmine, donepzil.
- Insecticide (organophosphate, carbamate)
- Antiseizure medications (carbamazepine, lacosamide, phenytoin).
- Opioids
- Sedative-hypnotics: Barbiturates, benzodiazepines.
- Other
◾️Metabolic
- Electrolyte disturbances: ⬆️⬇️K, ⬆️⬇️Ca, ⬆️Mg.
- Hyperkalemia: An increased likelihood of short-term adverse events was found for hyperkalemic patients whose ECG demonstrated QRS prolongation, bradycardia (HR<50), and/or junctional rhythm 4.
- BRASH Syndrome (Bradycardia, Renal failure, AV node blockade, Shock, and Hyperkalemia)
- Endocrine, metabolic, and environmental factors:
- Hypothyroidism.
- Adrenal insufficiency.
- Hypoglycemia.
- Uremia.
- Advanced liver disease.
- Hypothermia
- Severe hypoxia, e.g., obstructive sleep apnea, airway occlusion, abnormalities, or diseases that lead to respiratory depression.
- Hypercapnia / acidemia.
◾️Myocardial infarction
- Inferior MI: nodal ischemia and vagal response (self-limiting or responds to atropine)
- Anterior MI: infranodal ischemia (often requires pacing)
◾️Autonomic-mediated syndromes
- Carotid sinus stimulation or hypersensitivity
- Increased ICP (Cushing reflex)
- Vagal stimulation (coughing, micturition, defecation, vomiting)
- Vagal response e.g., after urinary catheter insertion for urinary retention 5
- Intra-abdominal hemorrhage
- Neurogenic shock
◾️Neurologic disorders
◾️Acute hypertension (sinus bradycardia).
◾️Infection
- Lyme disease, tertiary syphilis, and Chagas disease.
- Aortic valve endocarditis with ring abscess (conduction block)
- Encephalitis, dengue hemorrhagic fever, etc.
◾️Failure of a previously implanted pacemaker device.
◾️Other conditions *
- Genetic
-
Aging
- Idiopathic/senile sinus node dysfunction (usually occurs in elderly patients with idiopathic degeneration of the sinus node).
-
Athletes
- Myocarditis or pericarditis.
- Infiltrative disorders: Amyloid, sarcoid, hemochromatosis.
- Collagen vascular diseases, e.g., Systemic lupus erythematosus, scleroderma, rheumatoid arthritis.
- Conduction system injuries (e.g., surgery), trauma.
- Congenital heart disease
- Diving.
Clinical manifestation
Ranges from asymptomatic to shock, including impending cardiac arrest (figure 2).

The first step in the approach to the management of bradycardia is to recognize whether the patient’s hemodynamic status is stable or not.
By and large hemodynamic instability is defined as a myriad of hypotension and signs and symptoms of poor organ perfusion including altered mental status (AMS), ischemic chest pain, and dyspnea from acute heart failure. However hemodynamic instability is not all-or-none rather it has a spectrum.
- Occult bradycardic shock: In some patients with adequate physiologic reserve, compensatory vasoconstrictor response may maintain blood pressure and mental status despite the low cardiac output and poor organ perfusion (remember that BP is not equal to cardiac output). They require monitoring and urgent therapy, but they are not actively dying!
- Overt bradycardic shock: represents severe hemodynamic derangement with low BP and organ malfunction. These patients have worsening symptoms and deteriorating vital signs (e.g. HR< 30bpm or progressively worsening bradycardia) with imminent threat of cardiac arrest. Hence emergent resuscitation is warranted for these patients, the term bradycardic peri-arrest is a better term used for such a condition (aka impending cardiac arrest).
General approach to bradycardia in ED
◾️Fundamental concepts
The principles of management of bradycardia involve the following essential concepts:
- Always consider the major life-threatening causes of bradycardia and evaluate the patient throughout the resuscitation to identify and correct the primary problem (e.g. hyperkalemia, drug intoxication, myocardial infarction, and neurologic catastrophe).
- Remember that sinus bradycardia is a common pathway of impending death from any terminal disease state such as massive pulmonary embolism, septic shock etc.
- In patients with bradycardic peri-arrest with deteriorating vital signs focus your efforts on immediate resuscitation rather than reading the rhythm on EKG!
The general approach to bradycardia is shown in the following figure (figure 3)
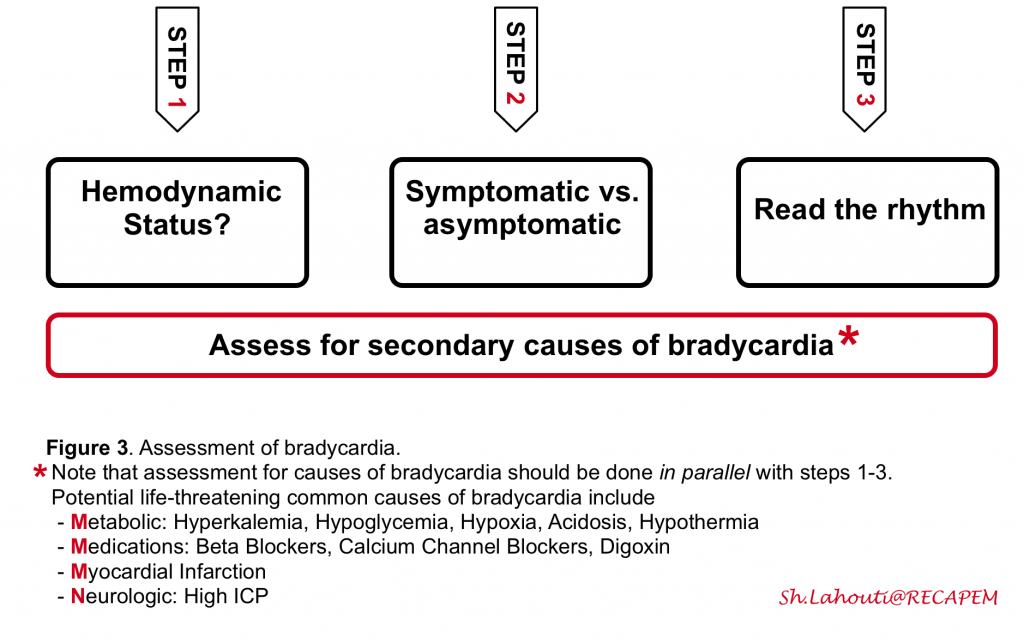
Step 1: Evaluate hemodynamic status (see above). In unstable bradycardic patients promptly start resuscitation.
Step 2: Determine if the bradycardia is causing symptoms (an older patient with underlying cardiac disease with chest pain and syncope), or if symptoms are the cause for bradycardia (e.g. vasovagal bradycardia).
Step 3: Read the rhythm. Correct identification of the location of the problem (SA node vs. AV node vs. His Purkinje) guides the management of bradycardia (whether urgent pacing is required or not). The simplified approach to Identifying the location of the problem involves:
- QRS width?
- Narrow QRS indicates the proximal (SA/AV node disease) site of the problem.
- Wide QRS indicates either proximal or distal (His Bundle disease) site of the disease. In this situation, assess rhythm before bradycardia:
- If sinus bradycardia → more likely to be proximal.
- If sinus tachycardia →more likely to be a distal disease.
Proximal problems (sinus and AV nodal dysfunction) rarely lead to life-threatening complications and are treated with watchful waiting, atropine, or sympathetic medications such as epinephrine and dopamine. However, the distal His-Purkinje block is much more serious and tends not to respond to atropine and sympathetic stimulation. These patients almost always need pacing and a definitive pacemaker.
👉Assess for potential life-threatening causes of bradycardia in parallel with the above 3 steps.
◾️The Structured Approach to Assessment of Bradycardia ⤵️
Download Bradycardia in ED (PDF)Evaluation
◾️History
- Obtaining a detailed history will assist in discovering the underlying etiology of the patient’s bradycardia:
- Renal impairment or failure suggestive of hyperkalemia
- A history of ischemic chest pain may suggest myocardial infarction
- Recent head trauma, headache, and altered mentation may suggest elevated ICP e.g. ICH, SAH.
- Blunt trauma, abdominal pain, distention, pregnancy (ectopic): Intra Abdominal hemorrhage
- Medication review
◾️Physical exam
- Hemodynamic stability: should be assessed first and adequacy of perfusion confirmed.
- Overt bradycardic shock: Low MAP; altered mental status in addition to other signs and symptoms of poor organ perfusion.
- Occult bradycardic shock: MAP and mental status intact, but cool extremities & poor urine output.
- Neuro/toxicologic exam
- Evidence of elevated intracranial pressure (e.g. stupor, widened optic nerve sheath)?
- 👉Patients with bradycardia can be drowsy or confused solely based on decreased cardiac output. However when mental status is disproportionately depressed for a given heart rate (e.g. GCS 8 in a patient with HR 45 bpm), one should consider neurologic causes of bradycardia like increased ICP and drug overdose.
- Pinpoint pupils may suggest toxic ingestion (e.g. clonidine or cholinergic agent)
- Evidence of elevated intracranial pressure (e.g. stupor, widened optic nerve sheath)?
◾️Bedside US: Cardiopulmonary and abdominal exam
- Volume status?
- Evidence of myocardial infarction (e.g. inferior wall motion abnormality)?
- Evidence of pulmonary congestion (e.g. B-lines throughout the lung fields)?
- Evidence of intra-abdominal free fluid?
◾️ECG: Focus on the following
- Signs of hyperkalemia (e.g. peaked T-waves, prolonged PR interval)
- Signs of ischemia; especially inferior MI.
- Rhythm diagnosis (e.g. sinus bradycardia vs. heart block)
- Deep symmetric TWI in anterior leads may suggest neurologic insults e.g. SAH
◾️Labs and imaging
- Fingerstick glucose
- Chemistries including Ca & Mg
- Troponin, if MI suggested by history/EKG
- Digoxin level, for patients taking digoxin
- Consider checking TSH
- Infectious titers (rapid plasma reagin for syphilis or Lyme)
- Complete blood count with cultures if an infection is suspected
- Coagulation panel
- Head CT scan to rule out increased intracranial pressure (ICP), if suspected of intracranial pathology (e.g. hemorrhage, mass, etc.)
- CT of the abdomen if there is concern for intra-abdominal hemorrhage.
Resuscitation
◾️Management of bradycardia is based on various factors, including
- Hemodynamic stability.
- Severity of symptoms.
- Risk of progression to third-degree AV block or asystole” *.
◾️Management of Stable but symptomatic patients with bradycardia is discussed here. In this post management of unstable bradycardia will be explored.
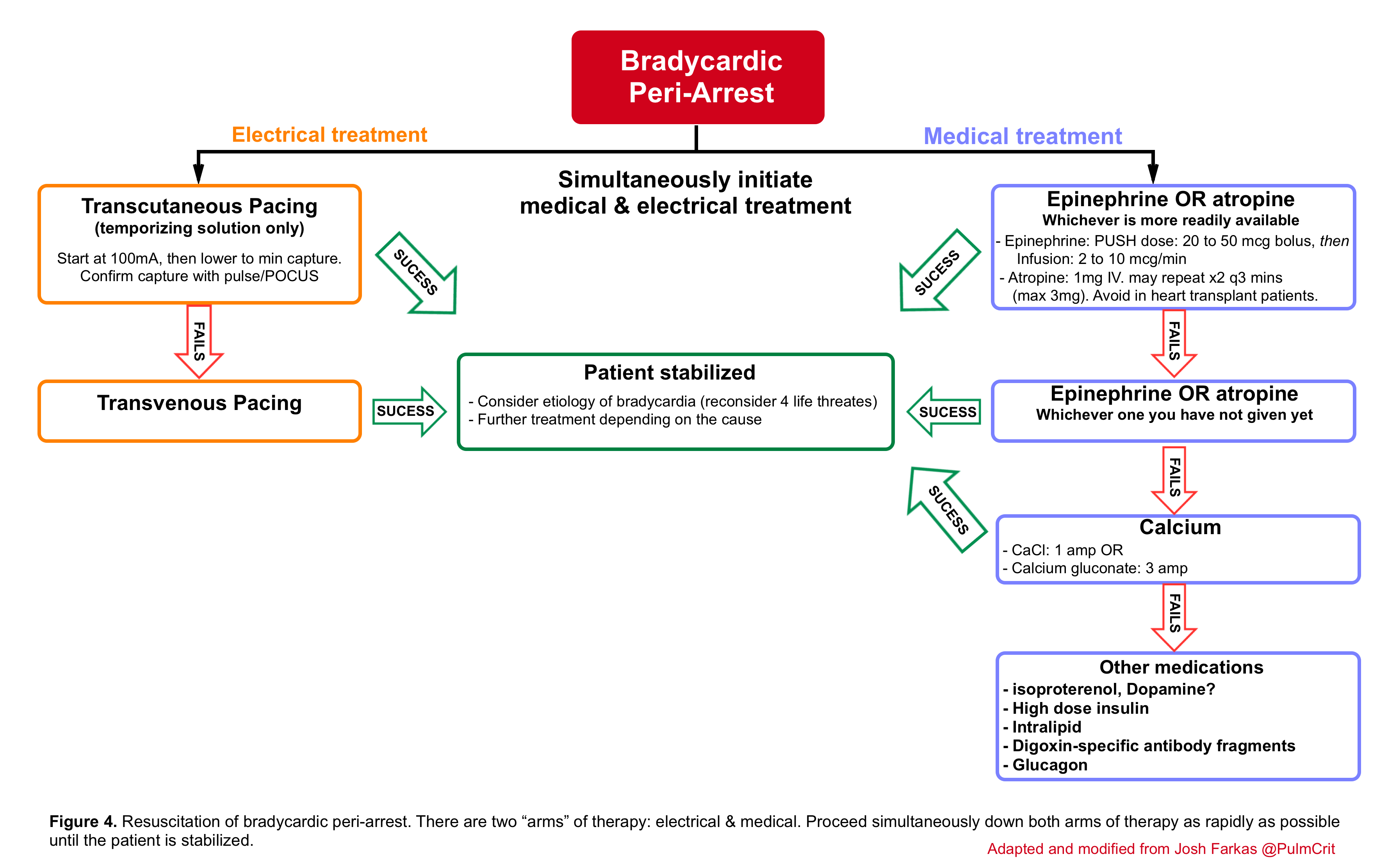
🟥Bradycardic peri-arrest may be defined as severe or progressive bradycardia with marked shock and concern for immediate cardiac arrest and therefore a maximally aggressive strategy should be chosen in order to prevent further deterioration into cardiac arrest (below algorithm).
- This involves the employment of two lines of treatment simultaneously; the medical (table 1) and electrical line.
- For patients with mild signs of organ malperfusion (e.g. normal blood pressure but poor urine output), then a more gradual and stepwise approach may be most appropriate. For example, simply starting an epinephrine infusion will often improve heart rate and perfusion.
Atropine
◾️Mechanism
- Atropine has a vagolytic effect that can affect sinus node automaticity, sinoatrial conduction, and AV conduction.
- Therefore it is effective only if the distal conduction system is conducting normally.
- Appropriately dosed atropine is usually effective for proximal AV block, sinus bradycardia, and junctional rhythms but is not useful (nor harmful) in distal AV block (idioventricular rhythms and second-degree type II and third-degree AV block).
◾️Caveats⚠️
- At low doses (< 0.5mg), atropine may cause paradoxical bradycardia 8
- At low doses, atropine decreases heart rate by blocking M1 acetylcholine receptors in the parasympathetic ganglion controlling the SA node.
- At higher doses, atropine increases heart rate by blocking M2 acetylcholine receptors in the myocardium itself.
- It is ineffective in heart transplant patients due to a lack of vagal innervation *.
- Studies have shown that atropine may precipitate paradoxical asystole in denervated hearts and therefore it is contraindicated to use in heart transplant patients 9.
- Atropine has a half-life of ~2 hours, and may stabilize the patient for 30-60 minutes, but then wear off *.
- This can initially make the patient appear stable, only to deteriorate later on, when everyone has stopped paying attention.
- Responsiveness to atropine in bradycardic patients is ~ 28% *.
- Therefore it is prudent not to delay other therapies while waiting for atropine to work.
◾️Dosing and strategy
- Strategy when atropine is used.
- Atropine is traditionally the first line of treatment, however for most unstable patients (Peri-arrest bradycardia) epinephrine is more effective and preferable.
- If atropine is the most immediately available drug, then it is administered, however, do not expect that it will fix everything.
- For very unstable bradycardia, give atropine while simultaneously preparing epinephrine and transcutaneous pacing.
- Atropine dosing
Epinephrine
Unlike atropine, epinephrine stimulates the entire myocardium. This provides epinephrine with a broader spectrum of efficacy for various mechanisms of bradycardia.
◾️Mechanism of hemodynamic stabilization
- Vasoconstriction via ⍺-1 receptor.
- Increased inotropy (contractility) and chronotropy (heart rate) via β-1 receptor.
- Epinephrine exerts its effects through α- and β-adrenergic receptors with dose-dependent actions
- At low doses (< 8-10 mcg/kg/min), the predominant effect is an inotrope and chronotrope with a minimal vasoconstrictive effect.
- At higher doses (>10mcg/kg/min) the vasoconstrictive effect (alpha-agonist effects) predominates.
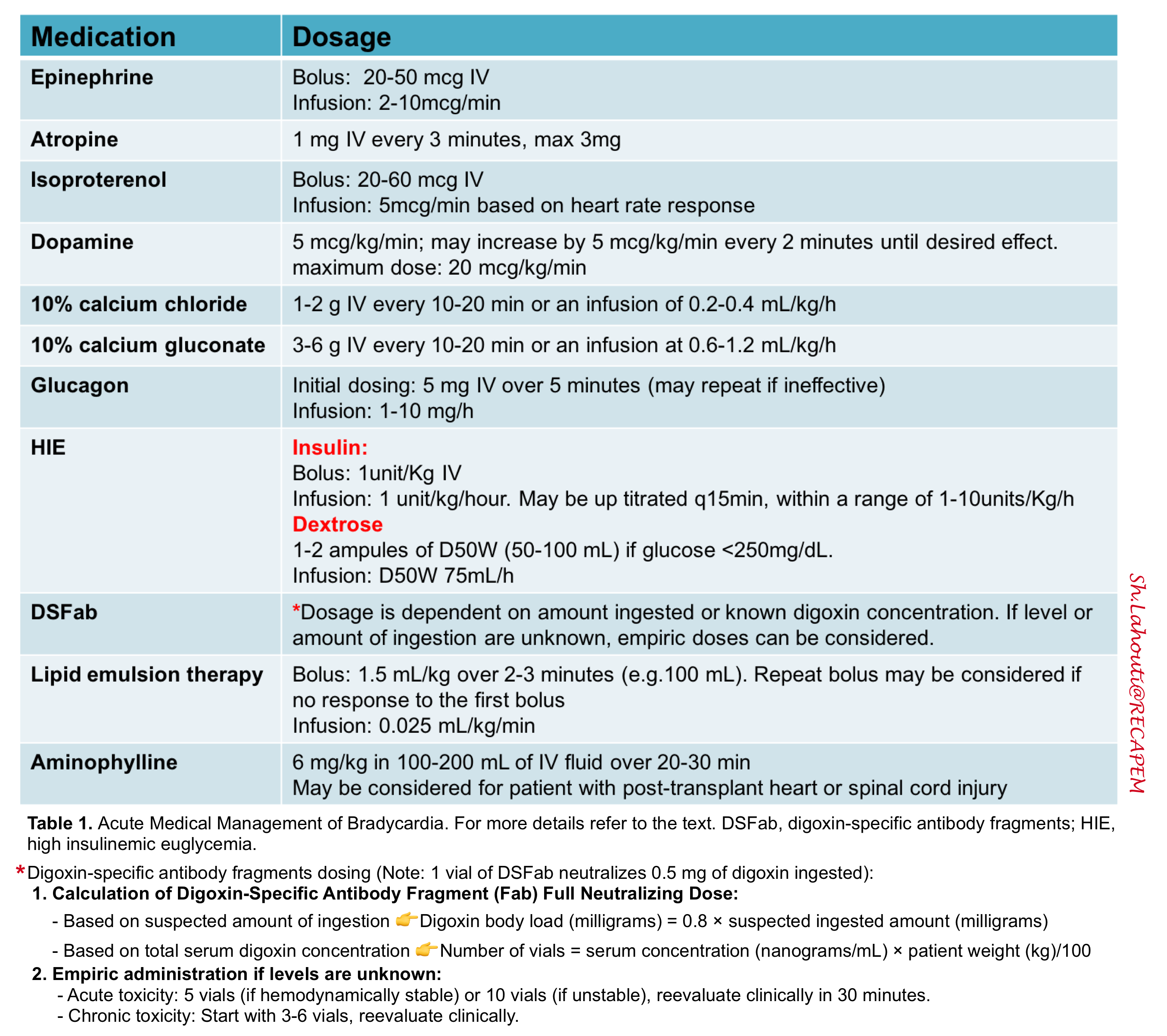
◾️Administration
- It is safe to use a proximal peripheral line initially with frequent limb checks. 12
- Preparation and use of epinephrine drip are shown below (Figure 5).
◾️Dosing
- Boluses for peri-arrest patient
- Boluses will stabilize the patient for a few minutes, but this is only a temporary bridge to an epinephrine infusion.
- Bolus doses ~ 20-50 mcg epinephrine.
- Epinephrine infusion
- The usual dose is 2-10 mcg/min (but there is no upper limit in a crashing patient) *.
- The dosing strategy depends on how unstable the patient is.
- For more unstable patients, start high and down-titrate as the patient responds.
- For patients who are fairly stable, start low and gradually up-titrate.
Calcium
◾️Consider IV calcium chloride or calcium gluconate; If atropine, epinephrine, and pacing are ineffective, and the cause of bradycardia is unclear.
◾️Calcium-responsive bradycardias
- Hyperkalemia
- Hypocalcemia
- Hypermagnesemia
- Calcium-channel blocker overdose
- Beta-blocker (may be effective)
◾️Dosing
- Bradycardia of unknown etiology: Can consider calcium chloride 1g or calcium gluconate 3g. 13
- Known or suspected Hyperkalemia: Calcium chloride 1g or calcium gluconate 3g. If ineffective and the patient is unstable, may repeat the dose once or twice if needed. 14
- Calcium channel blocker overdose:
Other medications
◾️Isoproterenol
- It is a pure beta-agonist (non-selective beta-adrenergic agonist).
- β1 Receptor Stimulation → ↑HR, ↑Contractility, ↑AV conduction
- β2 Receptor Stimulation (Vasculature & Bronchi) results in:
- Vasodilation → lowers peripheral vascular resistance (↓SVR). It can drop blood pressure, which may limit its use in hypotensive patients.
- Bronchodilation
- Some patients who don’t respond to epinephrine will respond to isoproterenol.
- Does not require central access for administration.
- Dosing: 20 to 60 mcg IV bolus, then 2-20 mcg/min IV infusion based on heart rate response *.
◾️Dopamine
- Traditionally used in symptomatic bradycardia.
- Mechanism
- It has a variety of receptors at different dose ranges.
- It is often difficult to figure out what dopamine is doing to the patient. For example, low-dose dopamine can actually cause hypotension (due to a predominant effect of vasodilation), which can make it difficult to wean off the dopamine.
- Disadvantages of dopamine compared to epinephrine:
- Dopamine can cause skin necrosis with prolonged infusion.
- Dopamine has a variety of different effects at different doses in different patients. This makes it difficult to titrate in any rational fashion (up-titration may cause dopamine to function via a different mechanism entirely).
- At high doses (>10-20 mcg/kg/min) may act predominantly as a vasoconstrictor (which may be an undesirable effect) and is also associated with tachydysrhythmias.
- Dopamine may directly stimulate diuresis via action on dopamine receptors, thereby falsely suggesting that renal perfusion is adequate 15.
- Dosing and strategy
- Strategy: If dopamine is the most readily available agent, then use it. When you have time, consider switching over to an epinephrine infusion.
- Dosing: 5 mcg/kg/minute; may increase by 5 mcg/kg/minute every 2 minutes until desired effect; maximum dose 20 mcg/kg/minute.
- ⚠️Dosages of >20 mcg/kg/min may result in vasoconstriction or arrhythmias *.
◾️Dobutamine
- Dobutamine is a β1-selective adrenergic agonist, with mild β2 and α1 effects (β1>β2>>α1) *.
- Less tachycardia relative to isoproterenol.
- Dobutamine tends to cause systemic vasodilation:
- Dobutamine might be perfect for a patient with bradycardia and normal/elevated blood pressure, where increasing cardiac output without increasing the blood pressure is aimed.
◾️Aminophylline
- Mechanism
- The suggested pharmacological effects of aminophylline include *:
- Inhibition of phosphodiesterase resulting in ↑intracellular cAMP (similar to milrinone).
- Direct and indirect effects on intracellular calcium concentration.
- Uncoupling of intracellular calcium.
- Antagonism of adenosine receptors.
- 💡Reminder. Mechanism of action of adenosine.
- Adenosine is an endogenous nucleotide that has a regulatory role in myocardial oxygen supply and demand mediated via A1 and A2a receptors.
- Activation of the A1-receptor results in slowing of the heart rate (negative chronotropic effect), slowing and blocking of AVN conduction (negative dromotropic effect), and antagonism of cardiac stimulatory effects of adrenergic agonists (anti-adrenergic effect).
- Activation of the A2a-receptor results in potent coronary artery vasodilation.
- Adenosine is an endogenous nucleotide that has a regulatory role in myocardial oxygen supply and demand mediated via A1 and A2a receptors.
- The half-life is 8 hours (but may be reduced to 4-5 hours with tobacco use).
- The suggested pharmacological effects of aminophylline include *:
- Indications
- Dosing
- Intravenous loading: The usual loading dose is 6 mg/kg ideal body weight, infused over 20-30 min.
- Maintenance therapy: Infusion of aminophylline (e.g. 0.3-0.5 mg/kg/h).
- Target level
- Chronotropy may be achieved at relatively low theophylline levels (5 ug/ml) *.
- It may also be reasonable to target a theophylline level of ~10-20 ug/ml (similar to its use in obstructive airway disease).
Therapy for Drug- or Toxin-related Bradycardia
◾️Glucagon
- Background
- It may be useful in patients with bradycardia and cardiac pump failure (especially those with beta-blockers rather than CCB intoxication) 18.
- It is unlikely to be helpful in patients with predominantly vasodilatory shock and CCB intoxication.
- Glucagon often induces vomiting so be cautious in patients with risk of aspiration.
- Glucagon should be reserved as a last resort and should not be given routinely.
- Glucagon test dose: Start with a loading dose of 5 mg IV over 5 minutes (may repeat if ineffective).
- Glucagon infusion: If the test dose causes hemodynamic improvement, this may be followed with a continuous infusion at a rate between 1-10 mg/hour, set equal to the dose of glucagon that caused clinical improvement (e.g., if 5 mg worked, set the infusion equal to 5 mg/hour).
- Glucagon has a half-life of about 15 minutes, so a continuous infusion is needed.
- Glucagon infusions may exhaust the hospital’s glucagon supply, so this may not be a sustainable long-term strategy. However, a glucagon infusion could be useful as a bridge for a few hours, until the high-dose insulin infusion starts to work.
◾️Digoxin-specific antibody fragments (DSFab)
- Digoxin toxicity can cause almost any bradyarrhythmias from junctional bradycardia to complete heart block19.
- DSFab is indicated (the best treatment) for hemodynamically significant bradyarrhythmias 20. More on this, here.
- Each vial contains 40mg of antibody fragments, which neutralize 0.5 mg of digoxin.
- The formula for calculating the number of vials required is (MDCalc: Digibind Dosing for Digoxin Poisoning):
- Chronic poisoning: The number of vials is estimated as (digoxin level in ng/ml)x(wt in kg)/100. However, lower doses may be considered initially, for patients with chronic digoxin toxicity who are clinically stable (e.g., initiate therapy with three vials and follow clinically to determine whether additional treatment is warranted) *
- Acute ingestion of known dose: Number of vials is estimated as (mg of digoxin ingested) x (1.6)*
- Empiric administration if levels are unknown *
- Acute toxicity: give 5 vials (if hemodynamically stable) or 10 vials (if unstable), and reevaluate clinically in 30 minutes.
- Chronic toxicity: start with 3-6 vials, and reevaluate clinically.
- ✅Atropine is a good temporizing measure (since patients with digoxin toxicity have excess vagal tone).
- ⚠️Avoid pacing or beta-agonists if possible, as these may provoke ventricular tachycardia 22.
◾️Lipid emulsion therapy (intralipid)
- Background
- It is the first line of treatment in patients suspected of local anesthetic systemic toxicity (LAST) 23. Suspect in any bradycardic patient on lidocaine infusion or recently treated with nerve block.
- Intralipid may also be considered for the following intoxications:
- Lipophilic β-blockers e.g. propranolol, carvedilol, penbutolol
- CCBs
- TCAs
- Local Anesthesia Systemic Toxicity (LAST)
- Dosing: New guidelines recommend a revised dosing scheme with a reduced maintenance infusion, which could optimize the risk/benefit balance. These recommend the following dosing scheme for 20% lipid emulsion (e.g. Intralipid) 24:
- Start with a bolus of 1.5 mL/kg over 2-3 minutes (e.g. 100 mL). Repeat bolus may be considered if there is no response to the first bolus.
- Give an additional 0.75 mL/kg over three minutes (e.g. 50 mL).
- Start a maintenance infusion rate of 0.025 mL/kg/min.
- If there is an initial response to the bolus followed by a re-emergence of instability during the maintenance infusion, consider re-bolusing and/or increasing the infusion rate. There is no known maximal dose, but it may be best to limit the dose to ~10 mL/kg/day total.
Transcutaneous pacing
In bradycardic peri-arrest, pacing should be started in parallel with medication (figure above). Transcutaneous pacing is a temporary measure until more definitive stabilization is possible (e.g. transvenous pacing).
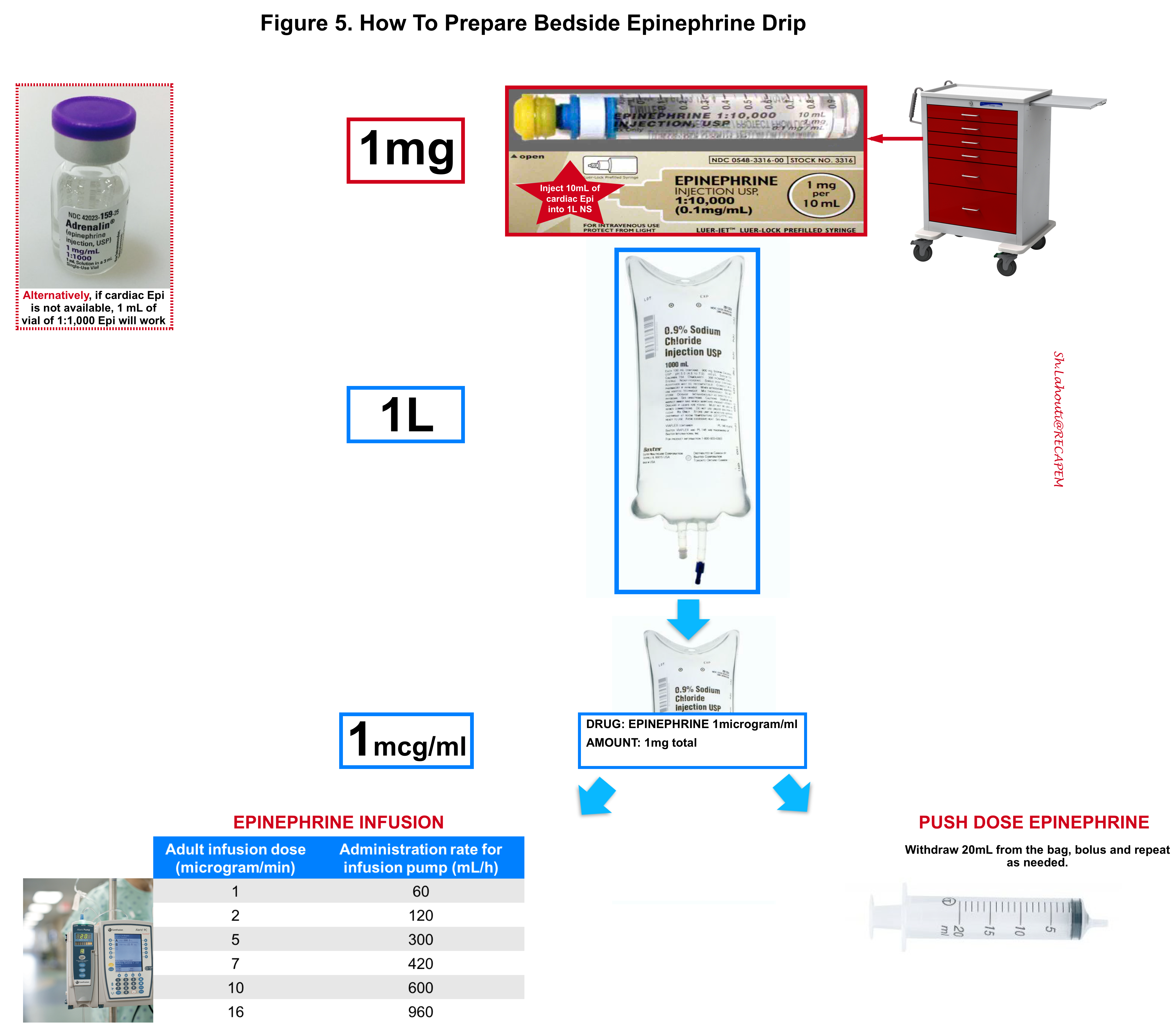
◾️Pad configuration
- There are 2 placement configurations recommended for optimal capture in transcutaneous pacing.
- Anterior Lateral: One pad is placed over the apex of the heart and the other one is placed on the right upper chest.
- Anterior Posterior placement: One pad is placed over the V3 lead position while the other is placed between the left scapula and the thoracic spine.
◾️Setting the output on the pacer
- Principles
- An initial pacing rate should be set at least as high as the intrinsic heart rate of the patient (although a standard rate of 80 bpm can be selected) with the current set to minimal output.
- Initially, pacer spikes may be visualized without resulting in cardiac depolarization. The current can be increased 5 to 10 mA at a time until a clear QRS complex and T wave are demonstrated following each pacer spike.
- Check the patient’s pulse at this point to confirm that the electrical “capture” has resulted in a mechanical response.
- This level is defined as the pacing threshold, and it will be found between 40 and 80 mA for most healthy patients.
- The final current output should be set to 5 to 10 mA above the threshold level to ensure continued capture.
- Patients with obesity and COPD typically require ~ 40-80 mA more than other patients to capture.
- ⚠️Be aware of pseudo-pacing: It is when the pacemaker isn’t capturing the myocardium, but the monitor displays a heart rate equal to the transcutaneous pacemaker.
- This provides a false sense of security because the monitor looks great.
- Always confirm real capture that results in a ventricular beat with femoral pulse checks (ideally using POCUS) and pulse oximetry wave.
- Do not rely solely on the monitor/ECG.
- Do not use a carotid pulse check for the assessment of circulation as TCP can create muscular movements that may feel like a carotid pulse.
- Assess circulation using the femoral pulse (with POCUS ideally).
◾️Analgesia/sedation
- Transcutaneous pacing can be quite uncomfortable for patients who are awake, as it requires the discharge of electrical impulses through the skin and chest wall muscles. Therefore, procedural sedation should be considered to reduce this discomfort.
- Consider Ketamine as your first-line analgesic for the patient undergoing transcutaneous pacing as it is least likely to cause hypotension, may help increase the heart rate, and helps maintain respirations
◾️Transcutaneous pacing in crashing patient
- Start at a maximal current (e.g. 100mA) and titrate downward to 5-20mA above the minimum energy required for capture; if not capturing, increase to max 130mA and if still not capturing, move the pads to improve the vector through the heart and try again.
◾️Pacing in a non-crashing patient: start low and titrate up.
- If the patient is doing ok, then you probably wouldn’t want to do transcutaneous pacing at all. However, it may be useful to determine if the patient responds to transcutaneous pacing. Proving that transcutaneous pacing will capture the heart may help you decide whether placing a transvenous pacemaker is necessary in a borderline patient.
Transvenous pacing
Transvenous is much more effective than transcutaneous pacing with success rates of >95%. Transvenous pacing is indicated as follows:
- Unstable bradycardia which doesn’t respond to other medication and/or interventions.
- High-degree AV blocks that leave the patient at ongoing risk of deterioration (e.g. Mobitz II, third-degree heart block with wide-complex escape rhythm).
RECAP:
- Do not rely solely on blood pressure to identify unstable patients. Keep in mind that BP is not equal to cardiac output. Some patients vasoconstrict and maintain normal blood pressure, despite organ malperfusion.
- For very unstable patients, start both medical and electrical arms of treatment until something works!
- Evaluate your patients throughout the resuscitation process for life-threatening common causes of bradycardia.
- Remember that bradycardia can be caused by myocardial infarction and various intoxications; so fixing the heart rate may not be enough to fix the patient. Reverse any other underlying cause of bradycardia.
- Don’t be fooled by transcutaneous pacemaker pseudocapture. The fact that the chest is twitching and the monitor shows a normal heart rate means nothing; it’s still possible that the myocardium isn’t being captured.
- Pacing is unlikely to be successful in B-blocker, Ca-blocker, and digoxin poisonings.
Going further
- EM:RAP: C3 Bradycardia- Initial approach
- Managing unstable bradycardia (First10em)
- Bradycardia (FOAMcast)
- Bradycardia (IBCC)
Post Peer Reviewed By: Mojtaba Chardoli. MD.
Reference
1. Kadish AH, Buxton AE, Kennedy HL, Knight BP, Mason JW, Schuger CD, Tracy CM, Boone AW, Elnicki M, Hirshfeld JW Jr, Lorell BH, Rodgers GP, Tracy CM, Weitz HH. ACC/AHA clinical competence statement on electrocardiography and ambulatory electrocardiography. A report of the ACC/AHA/ACP-ASIM Task Force on Clinical Competence (ACC/AHA Committee to Develop a Clinical Competence Statement on Electrocardiography and Ambulatory Electrocardiography). J Am Coll Cardiol. 2001 Dec;38(7):2091-100. doi: 10.1016/s0735-1097(01)01680-1. PMID: 11738321
2. Wung SF. Bradyarrhythmias: Clinical Presentation, Diagnosis, and Management. Crit Care Nurs Clin North Am. 2016 Sep;28(3):297-308. doi: 10.1016/j.cnc.2016.04.003. Epub 2016 Jun 22. PMID: 27484658
3. Deal N. Evaluation and management of bradydysrhythmias in the emergency department. Emerg Med Pract. 2013 Sep;15(9):1-15; quiz 15-6. Epub 2013 Aug 10. PMID: 24044868
4. Durfey N, Lehnhof B, Bergeson A, Durfey SNM, Leytin V, McAteer K, Schwam E, Valiquet J. Severe Hyperkalemia: Can the Electrocardiogram Risk Stratify for Short-term Adverse Events? West J Emerg Med. 2017 Aug;18(5):963-971. doi: 10.5811/westjem.2017.6.33033. Epub 2017 Jul 10. PMID: 28874951; PMCID: PMC5576635.
5. Seifert PC, Yang Z, Reines HD. Crisis Management of Unstable Bradycardia in the OR. AORN J. 2016 Feb;103(2):215-23. doi: 10.1016/j.aorn.2015.12.012. PMID: 26849986.
6. Wung SF. Bradyarrhythmias: Clinical Presentation, Diagnosis, and Management. Crit Care Nurs Clin North Am. 2016 Sep;28(3):297-308. doi: 10.1016/j.cnc.2016.04.003. Epub 2016 Jun 22. PMID: 27484658
7. Cortes J, Hall B, Redden D. Profound symptomatic bradycardia requiring transvenous pacing after a single dose of tizanidine. J Emerg Med. 2015 Apr;48(4):458-60. doi: 10.1016/j.jemermed.2014.10.005. Epub 2014 Nov 20. PMID: 25456780
8. Chin KJ, Seow SC. Atrioventricular conduction block induced by low-dose atropine. Anaesthesia. 2005 Sep;60(9):935-6. doi: 10.1111/j.1365-2044.2005.04346.x. PMID: 16115264
9. Bernheim A, Fatio R, Kiowski W, Weilenmann D, Rickli H, Brunner-La Rocca HP. Atropine often results in complete atrioventricular block or sinus arrest after cardiac transplantation: an unpredictable and dose-independent phenomenon. Transplantation. 2004 Apr 27;77(8):1181-5. doi: 10.1097/01.tp.0000122416.70287.d9. PMID: 15114081
10. Brady WJ, Swart G, DeBehnke DJ, Ma OJ, Aufderheide TP. The efficacy of atropine in the treatment of hemodynamically unstable bradycardia and atrioventricular block: prehospital and emergency department considerations. Resuscitation. 1999 Jun;41(1):47-55. doi: 10.1016/s0300-9572(99)00032-5. PMID: 10459592
11. Eddleston M, Buckley NA, Eyer P, Dawson AH. Management of acute organophosphorus pesticide poisoning. Lancet. 2008 Feb 16;371(9612):597-607. doi: 10.1016/S0140-6736(07)61202-1. PMID: 17706760; PMCID: PMC2493390.
12. Le A, Patel S. Extravasation of Noncytotoxic Drugs: A Review of the Literature. Ann Pharmacother. 2014 Jul;48(7):870-886. doi: 10.1177/1060028014527820. Epub 2014 Apr 8. PMID: 24714850
13. Lavonas EJ, Drennan IR, Gabrielli A, Heffner AC, Hoyte CO, Orkin AM, Sawyer KN, Donnino MW. Part 10: Special Circumstances of Resuscitation: 2015 American Heart Association Guidelines Update for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2015 Nov 3;132(18 Suppl 2):S501-18. doi: 10.1161/CIR.0000000000000264. Erratum in: Circulation. 2016 Aug 30;134(9):e122. PMID: 26472998.
14. Dépret F, Peacock WF, Liu KD, Rafique Z, Rossignol P, Legrand M. Management of hyperkalemia in the acutely ill patient. Ann Intensive Care. 2019;9(1):32. Published 2019 Feb 28. doi:10.1186/s13613-019-0509-8
15. Overgaard CB, Dzavík V. Inotropes and vasopressors: review of physiology and clinical use in cardiovascular disease. Circulation. 2008 Sep 2;118(10):1047-56. doi: 10.1161/CIRCULATIONAHA.107.728840. PMID: 18765387.
16. Woodward C, Pourmand A, Mazer-Amirshahi M. High dose insulin therapy, an evidence based approach to beta blocker/calcium channel blocker toxicity. Daru. 2014 Apr 8;22(1):36. doi: 10.1186/2008-2231-22-36. PMID: 24713415; PMCID: PMC3985540.
17. Engebretsen KM, Kaczmarek KM, Morgan J, Holger JS. High-dose insulin therapy in beta-blocker and calcium channel-blocker poisoning. Clin Toxicol (Phila). 2011 Apr;49(4):277-83. doi: 10.3109/15563650.2011.582471. PMID: 21563902.
18. Graudins A, Lee HM, Druda D. Calcium channel antagonist and beta-blocker overdose: antidotes and adjunct therapies. Br J Clin Pharmacol. 2016 Mar;81(3):453-61. doi: 10.1111/bcp.12763. Epub 2015 Oct 30. PMID: 26344579; PMCID: PMC4767195
19. Ma G, Brady WJ, Pollack M, Chan TC. Electrocardiographic manifestations: digitalis toxicity. J Emerg Med. 2001 Feb;20(2):145-52. doi: 10.1016/s0736-4679(00)00312-7. PMID: 11207409.
20. Chan BS, Isbister GK, O’Leary M, Chiew A, Buckley NA. Efficacy and effectiveness of anti-digoxin antibodies in chronic digoxin poisonings from the DORA study (ATOM-1). Clin Toxicol (Phila). 2016 Jul;54(6):488-94. doi: 10.1080/15563650.2016.1175620. Epub 2016 Apr 27. PMID: 27118413.
21. Chan BS, Buckley NA. Digoxin-specific antibody fragments in the treatment of digoxin toxicity. Clin Toxicol (Phila). 2014 Sep-Oct;52(8):824-36. doi: 10.3109/15563650.2014.943907. Epub 2014 Aug 4. PMID: 25089630
22. Kanji S, MacLean RD. Cardiac glycoside toxicity: more than 200 years and counting. Crit Care Clin. 2012 Oct;28(4):527-35. doi: 10.1016/j.ccc.2012.07.005. Epub 2012 Aug 30. PMID: 22998989.
23. El-Boghdadly K, Pawa A, Chin KJ. Local anesthetic systemic toxicity: current perspectives. Local Reg Anesth. 2018 Aug 8;11:35-44. doi: 10.2147/LRA.S154512. PMID: 30122981; PMCID: PMC6087022.
24. American College of Medical Toxicology. ACMT Position Statement: Guidance for the Use of Intravenous Lipid Emulsion [published correction appears in J Med Toxicol. 2016 Dec;12 (4):416]. J Med Toxicol. 2017;13(1):124-125. doi:10.1007/s13181-016-0550-z
Add comment