

20 October 2023 by Shahriar Lahouti.
CONTENTS
- Preface
- Mechanism of action and pathophysiology
- Cardiomyocyte
- Autonomic nervous system
- Myocardial conduction tissue
- Toxicokinetics
- Risk stratification
- Signs and symptoms
- Clinical toxidrome
- Differential diagnosis
- Diagnosis
- Treatment
- Specific conditions
- Rapid review
- Media
- Going further
- References
Preface
Digoxin has a narrow therapeutic index and toxicity is common. It continues to be a common medication utilized by patients presenting to the ED, however, nonspecific signs and symptoms make the diagnosis of digoxin toxicity difficult to establish, and serum digoxin concentrations do not always correlate with toxicity. The key to successful management is prompt diagnosis and early identification of those patients who benefit from digoxin-specific antibody fragments (DSFab).
Mechanism of action and pathophysiology
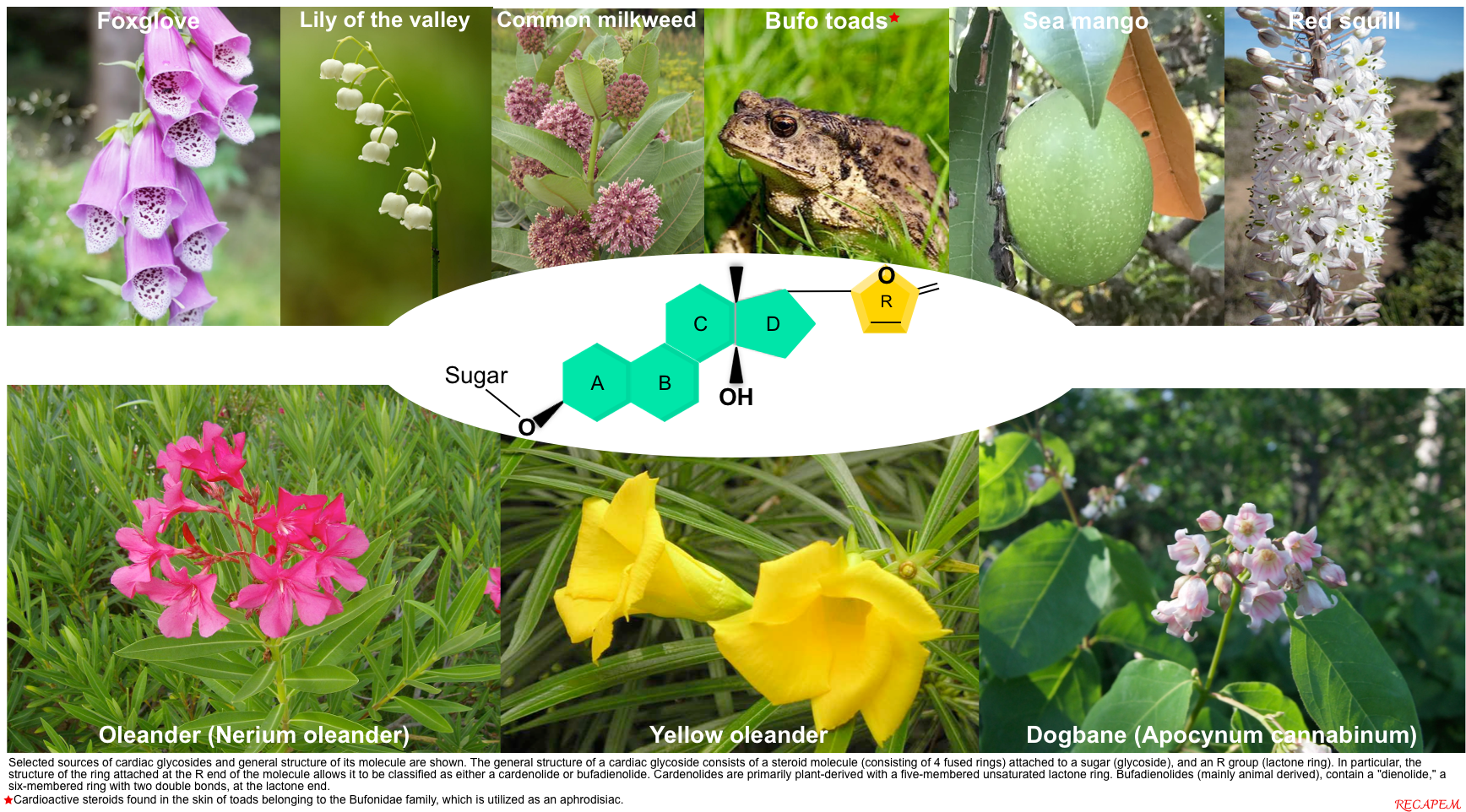
In addition to digoxin, other naturally occurring cardiac glycosides exist, including foxglove, oleander, yellow oleander, red squill, lily of the valley, and dogbane. Similarly, bufadienolide is a cardioactive steroid found in the skin of Bufo toads, which is utilized as an aphrodisiac *. While the individual toxins differ slightly among each of these sources, all are cardiac glycosides and, as such, exhibit similar toxicity.
Effect on cardiomyocyte (positive inotropy)
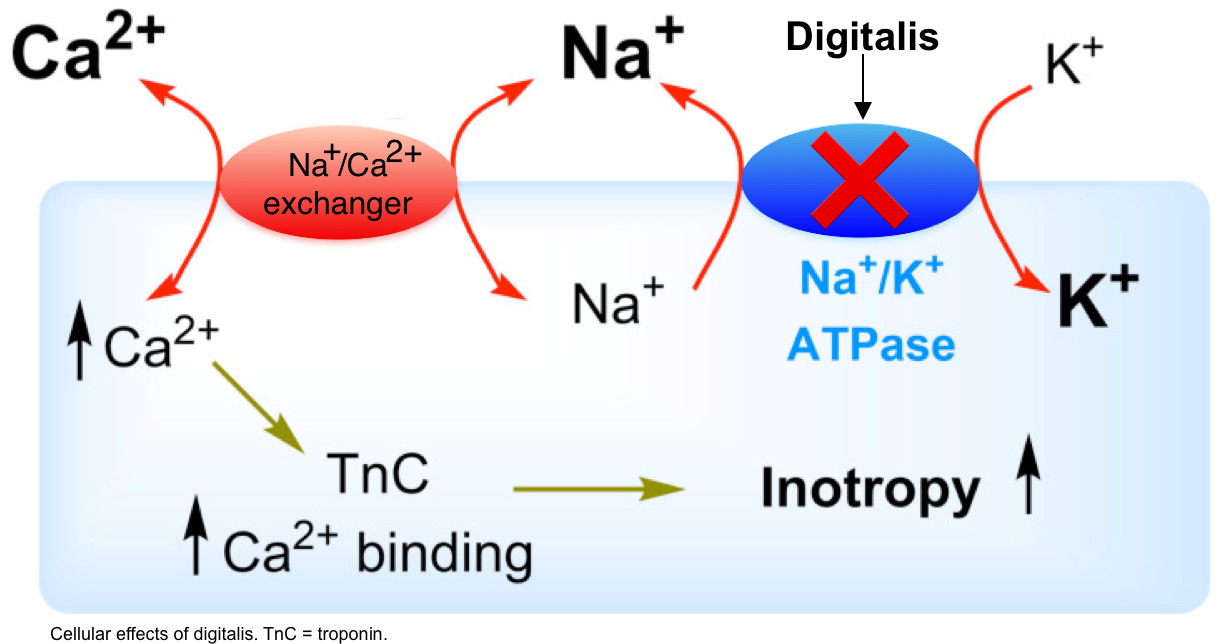
- Cardiac glycosides reversibly inhibit sodium-potassium-ATPase *. This causes an increase in intracellular sodium and a decrease in intracellular potassium (figure below).
- The increase in intracellular sodium promotes the activity of the sodium-calcium exchanger, increasing intracellular calcium concentration.
- Increased intracellular calcium increases inotropy.
- The fluxes in calcium can also have electrophysiologic effects (discussed below).
Effects on the autonomic nervous system
- Digoxin binds to Na/K ATPase in nerve terminals which causes augmentation of intracellular Ca2+. This will cause depolarization of efferent nerve terminals and release of neurotransmitters.
- Cardiac glycosides increase vagal tone via action at the carotid body *, thereby reducing conduction through the sinoatrial and atrioventricular nodes *,*.
- The parasympathetic system is affected by CASs by an increase in the release of acetylcholine from vagal fibers.
- In toxic concentrations, cardiac glycosides can increase sympathetic tone *.
Effect on myocardial conducting tissue
Basic electrophysiology: Action potential
Normal depolarization of the cardiac myocyte and pacemaker cells is shown below.
- In cardiac myocytes, depolarization begins with the opening of the fast sodium channels (phase 0).
- The resulting increase in intracellular sodium, and subsequent change in the resting membrane potential, opens voltage-gated calcium channels (phase 2).
- Repolarization of cardiocytes is mediated through efflux of potassium (phase 3).
- Sodium is then removed from the cell by the sodium-potassium-ATPase (phase 4). Some calcium is removed from the cell by the sodium-calcium antiporter.
Normal depolarization of pacemaker cells by influx of calcium through L-type calcium channels (phase 0). This is followed by repolarization (phase 3) which occurs by the efflux of potassium.
- In pacemaker cells, the slope of phase 4 is called pacemaker potential, which is affected by various channels, some of which are shown below.
- For example, activation of muscarinic receptors can cause hyperpolarization of cells with negative chronotropic and dromotropic effects. Epinephrine and norepinephrine have opposite effects (i.e. fastening depolarization which could be mediated through activation of the HCN channel, shown below).
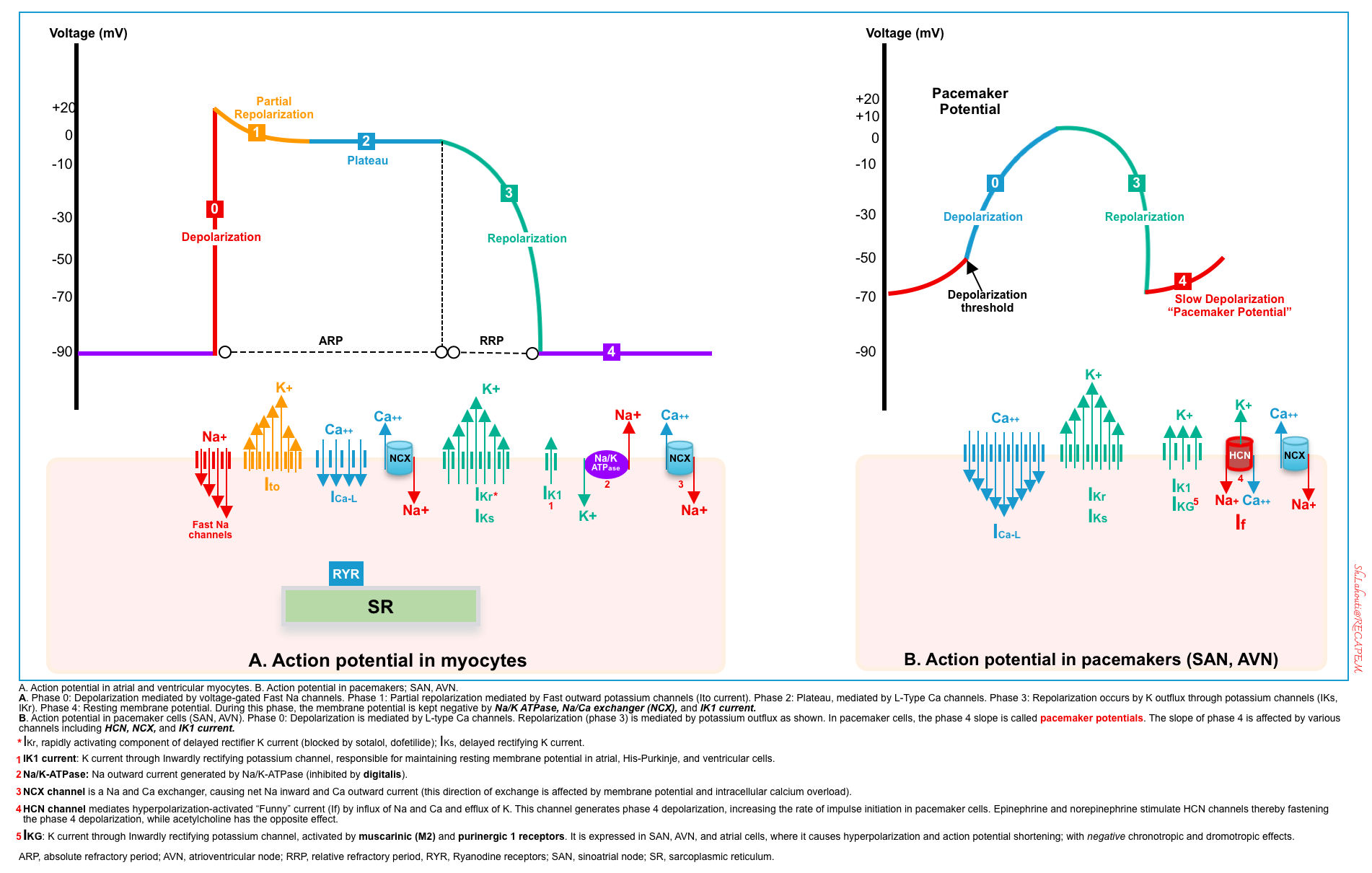
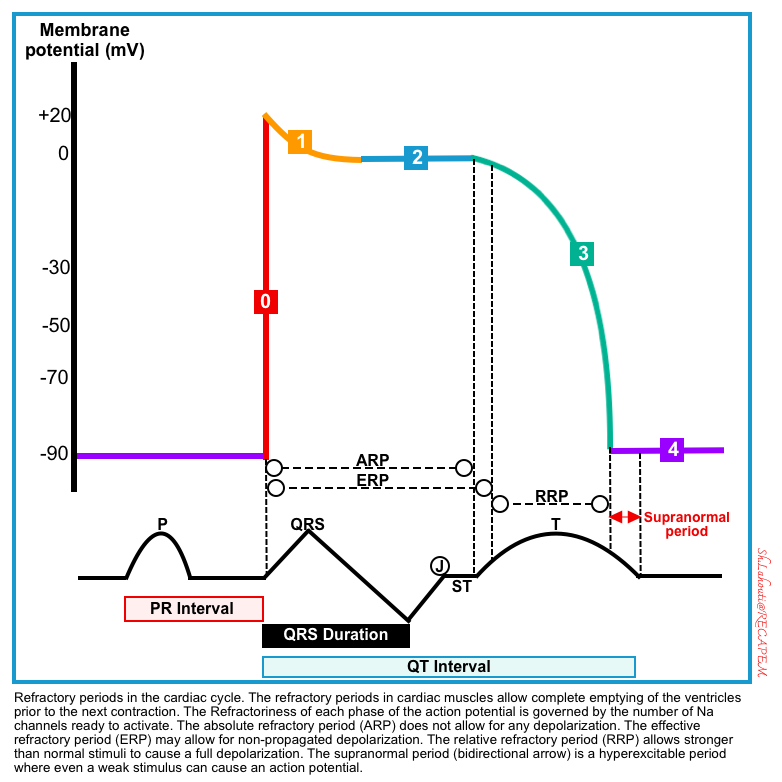
Refractory periods
Cardiac cells have 3 refractory periods as shown below. The refractoriness of each is governed by the number of sodium channels ready to activate.
- Absolute refractory period (from the beginning of phase 0 till the end of phase 2). No depolarization is allowed during this period.
- Effective refractory period (from the beginning of phase 0 to the very initial part of phase 3). Only non-propagated depolarizations might be allowed.
- Relative refractory period (from the very initial part of phase 3 to the beginning of phase 4). This period will allow full depolarization to those stimuli that are stronger than normal stimuli.
Digitalis effect on electrophysiology
The digitalis effect on myocardial conduction tissue is illustrated below *. These include:
- Slowing phase 0 leads to reduced conduction velocity. Reduced conduction velocity in AVN is manifested as an increased PR interval
- Shortening phase 2 causes more rapid repolarization manifested as QT interval shortening and ST-segment and T-wave forces opposite in direction to the major QRS forces on ECG.
- Increasing the slope of phase 4 which leads to enhanced automaticity.
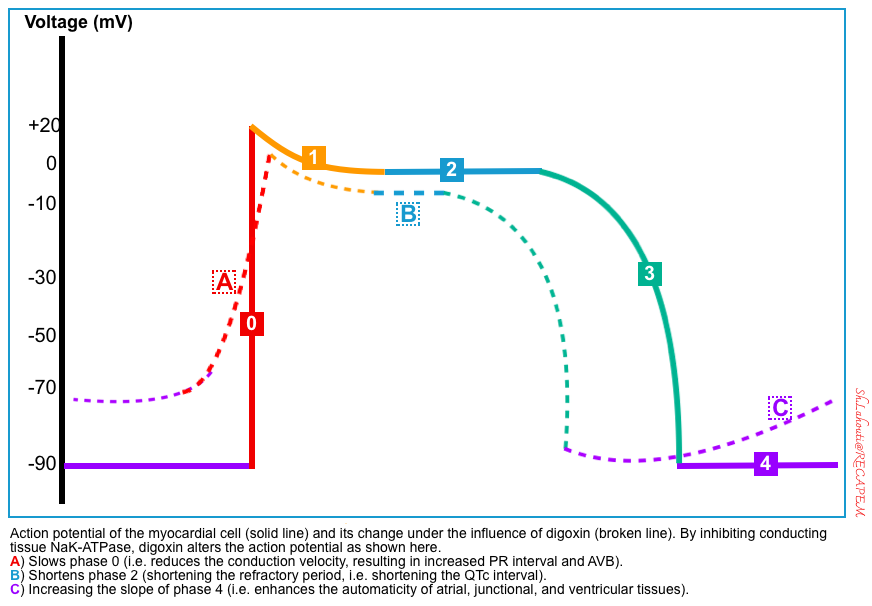
Reduced conduction velocity (negative dromotrophy)
- Digoxin inhibits the Na/K ATPase pump resulting in more positive resting potentials (e.g. -90 mV to -70 mV).
- At a more positive resting potential, the voltage-dependent fast Na channels are inhibited. Therefore, the amplitude of phase 0 is reduced (↓conduction velocity).
- Reduced conduction velocity in AVN is manifested as increased PR interval and AVB.
Shortened atrial and ventricular refractory period
- Digoxin causes elevated intracellular calcium as explained above. This has the following electrophysiologic effects:
- High intracellular Ca will inhibit L-type Ca channels, decreasing inward Ca current during the plateau phase.
- High intracellular calcium causes stimulation of calcium-dependent K channels during phase 3. This will lead to more rapid efflux of K which causes more repaid repolarization.
- These two mechanisms cause more rapid repolarization of atrial and ventricular myocytes (shortened repolarization intervals).
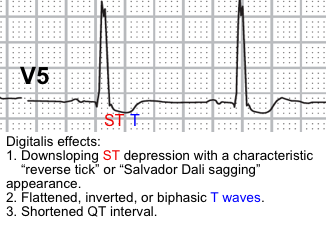
- These manifest on ECG as QT interval shortening and ST-segment and T-wave forces opposite in direction to the major QRS forces. The last effect results in the characteristic scooping of the ST segments (referred to as the digitalis effect). More on this: Digitalis (Dr.Smith’s ECG Blog)
Sinoatrial node (SAN) and atrioventricular node
- The rate of depolarization and conduction are concurrently decreased in SAN and AVN. This is medicated by enhancement in vagally mediated parasympathetic tone.
- These changes in nodal conduction are reflected on the electrocardiogram (ECG) by a decrease in ventricular response rate to suprajunctional rhythms (e.g. slow ventricular response in patients with atrial fibrillation) and by PR interval prolongation.
Enhanced automaticity
- Excessive increases in intracellular Ca can result in delayed afterdepolarizations (DADs), which may in turn lead to premature contractions and trigger dysrhythmias.
- Cardiac glycosides shorten repolarization of the atria and ventricles, decreasing the refractory period of the myocardium, thereby increasing automaticity and the risk for dysrhythmias.
| Atria and ventricles | AV node | Electrophysiologic findings | |
| Excitability | ⬆️ | – | Extrasystoles, Tachydysrhythmias |
| Automaticity | ⬆️ | – | Extrasystoles, Tachydysrhythmias |
| Conduction velocity | ⬇️ | ⬇️ | ⬆️PR interval, AV block |
| Refractoriness | ⬇️ | ⬆️ | ⬆️PR interval, AV block ⬇️QT interval |
Toxicokinetics
Digoxin is available in multiple formulations, each differing slightly in bioavailability (table below).
- 25% of digoxin is bound to plasma proteins, mainly albumin. and has a large volume of distribution.
- Digoxin is mostly (~70%) excreted unchanged into urine. P-glycoprotein, an efflux pump, mediates the excretion of digoxin and many other drugs into urine.
- P-glycoprotein inhibitors (e.g. verapamil) will increase digoxin levels, while P-glycoprotein inducers (e.g. rifampin) will decrease digoxin levels.
- The half-life and time to steady-state varies by patient and is dependent on the renal function.
- Digoxin has a very narrow therapeutic index.
| Pharmacokinetic properties | Digoxin (injection) | Considerations |
| Bioavailability | Injection: 100% Capsule: 90% Oral elixir: 80% Oral tablet: 70% | |
| Plasma binding (percent) | 25 | Reduced protein binding of digoxin→↑ free serum digoxin levels: 🔸Warfarin 🔸Phenobarbital |
| Volume of Distribution (L/kg) | 5–7 (adults) 4–5 (adults with kidney failure) | Vd is higher in infants, neonates, elderly, and obese. |
| Onset of action | Injection: 5–30 min Oral: 0.5-2 h | |
| Peak effect | IV:1–4 h Oral:2-6 h | |
| Metabolism | Undergoes hydrolysis, oxidation, and conjugation. Metabolism is not dependent on the CYP-450 system. | |
| Excretion | First order kinetics Route: Renal (%70 unchanged), with limited hepatic metabolism Digoxin is secreted into the urine by P-glycoprotein* Not removed by dialysis. | P-glycoprotein inhibitors will increase digoxin levels: 🔸Azole antifungals. 🔸Azithromycin, erythromycin, clarithromycin. 🔸Amiodarone, carvedilol, ranolazine, ticagrelor. 🔸Verapamil, tacrolimus, cyclosporine. P-glycoprotein inducers will decrease digoxin levels 🔹Carbamazepine, fosphenytoin, phenobarbital, rifampin. |
| Half-life | Healthy adult patients: 1.5-2 d | Anuric adult patients: 3.5-5 d |
| Desired therapeutic concentrations | 0.5-0.9 ng/ml * (reference range is 0.8 to 2.0 ng/mL). | SDC doesn’t correlate well with clinical toxicity. Levels associated with severe toxicity: 🔸Acute intoxication: >10 ng/ml. 🔸Chronic intoxication: >4 ng/ml |
| Time to steady state | Healthy adult patients: 5-7 d | Anuric adult patients:15-20 d |
Risk stratification
- High total body load of digoxin
- Acute ingestion of digoxin > 10mg.
- Serum digoxin level of >10 ng/ml in acute intoxication, or >4ng/ml in chronic intoxication. More on this, below.
- Polypharmacy
- Altered pharmacokinetic
- P-glycoprotein inhibitors (table above).
- Other medications that cause increased digoxin levels (through different mechanisms) include alprazolam, atorvastatin, diphenoxylate, NSAIDs, spironolactone, warfarin, phenobarbital, and tetracycline *.
- Enhanced pharmacodynamic effects: Concomitant drug that potentiates digoxin toxicity.
- Beta-blocker, and nondihydropyridine calcium channel blocker: May result in advanced or complete heart block.
- Thiazide or loop diuretics: Electrolyte disturbances (⬇️K, ⬇️Mg) may lead to arrhythmia.
- Sympathomimetics: Arrhythmia
- Succinylcholine: Arrhythmia
- Altered pharmacokinetic
- Hypothyroidism *
- Age > 65 years *
- Elderly patients often have decreased in muscle mass resulting in a low lean body weight *. Elderly patients may also have decreased renal function, which could slow the rate of digoxin clearance from the body.
- Heart failure *
- Renal insufficiency
- Dehydration
- Liver disease
- Comorbid conditions: Toxic manifestations occur at substantially increasing frequency as the plasma rises above 2.0 ng/mL, however, signs of toxicity may occur at levels < 1.5 ng/mL in the presence of one or more comorbid conditions *:
- Electrolyte disturbance: ⬇️K, ⬇️Mg, ⬆️Ca, ⬆️Na.
- Hypokalemia: When potassium is low, more K-binding sites are open for digoxin binding (on Na/K-ATPase), increasing the effective concentration within the heart. Severe hypokalemia (<2.5 mEq/L) reduces the efficacy of Na/K pump function, slowing the pump and sensitizing Na/K-ATPase to the inhibitory effect of digoxin.
- Hypomagnesemia: Magnesium is a cofactor of the Na/K ATPase exchange. Hypomagnesemia will have the same inhibitory effects on the Na/K pump just like hypokalemia.
- Hypoxemia (especially in the setting of acute respiratory failure) *.
- Myocardial ischemia *.
- Electrolyte disturbance: ⬇️K, ⬇️Mg, ⬆️Ca, ⬆️Na.
Signs and symptoms of digoxin toxicity
Cardiovascular
- Cardiac glycoside toxicity can produce a range of cardiac dysrhythmias, and rhythm disturbances may evolve and change rapidly.
- There are no pathognomonic ECG findings, but the most classic dysrhythmias for digoxin intoxication include those dysrhythmias with the presence of both digoxin arrhythmogenic effects (vagotonic and enhanced automaticity) including *:
- Paroxysmal atrial tachycardia with AV block
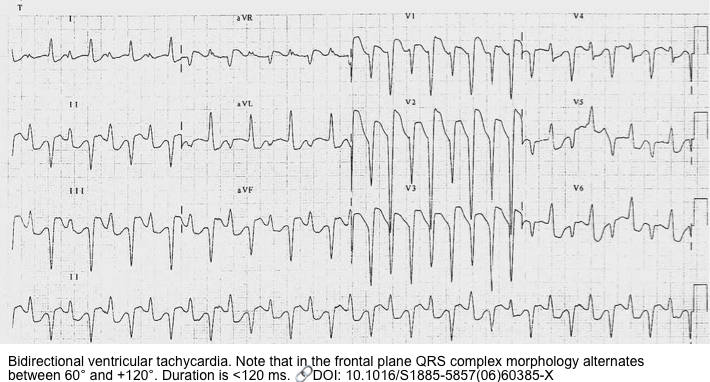
- Bidirectional ventricular tachycardia
- Mechanism: This rhythm is caused by junctional escape foci, but when traveling downstream to the ventricles, it passes alternatively through the right bundle and then the left bundle branch, giving rise to a bidirectional appearance.
- Bidirectional VT is not specific for digoxin toxicity and it can be seen in other conditions including familial catecholaminergic polymorphic VT *.
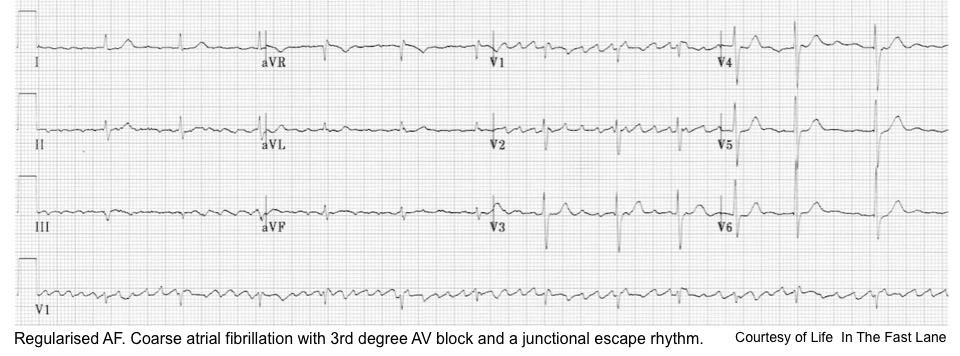
- Atrial fibrillation (with slow ventricular response, or regularized AF *) and a junctional escape rhythm *
- Junctional tachycardia *.
- AV junctional escape rhythms result when digitalis suppresses impulse formation at the sinoatrial node to the degree that the inherent AV pacemaker cells outpace the SA nodal cells. The escape rhythm usually manifests with a regular ventricular rate of 40 – 60 beats per minute but accelerated junctional rhythms are common, often with varying degrees of block.
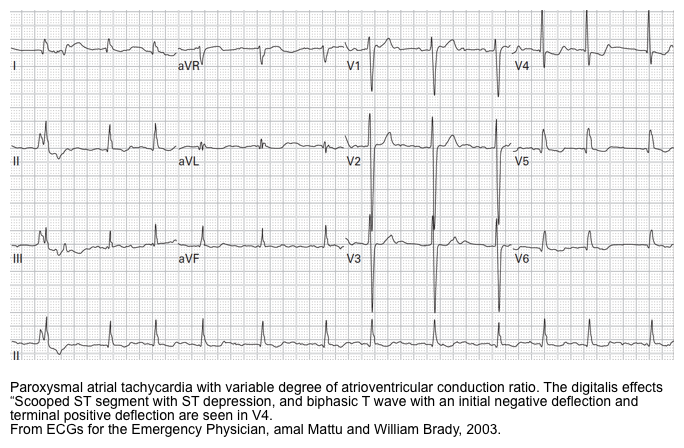
- Other rhythm disturbances *
- The most common rhythm disturbance is premature ventricular contractions (including ventricular bigeminy and trigeminy). These might be the earliest dysrhythmia.
- Sinus bradycardia or AV block
- AVB is more likely to develop in patients with atrial fibrillation or flutter. This is due to the combined effect of rapid atrial impulse and digoxin intoxication on AVN.
- Conduction block in this setting can be of any degree from first-degree AV block to complete heart block, though Mobitz type II is considered rare *.
- Atrial fibrillation with slow ventricular rate, or regularized AF.
- Ventricular tachycardia, ventricular fibrillation.
GI
- Anorexia, nausea, vomiting
- Diarrhea
- Abdominal pain
Neurologic
- Altered mental status: Lethargy, delirium, confusion, and disorientation can develop independent of hemodynamic parameters upon distribution of the drug into CNS.
- Fatigue, weakness (independent of hemodynamic parameters)
- Seizure (rare)
- Visual disturbance: Altered color perception (chromatopsia), blurred vision, photophobia, diplopia, blindness) *.
- Chromatopsia, specifically xanthopsia (objects appearing yellow), is classically associated with cardiac glycoside toxicity but is frequently absent and
not necessary for diagnosis.
- Chromatopsia, specifically xanthopsia (objects appearing yellow), is classically associated with cardiac glycoside toxicity but is frequently absent and
Endocrine/metabolic
- Hyperkalemia is caused by the inhibition of the sodium-potassium-ATPase in the skeletal muscle (as skeletal muscle is the largest reservoir for potassium in the body).
Clinical toxidrome
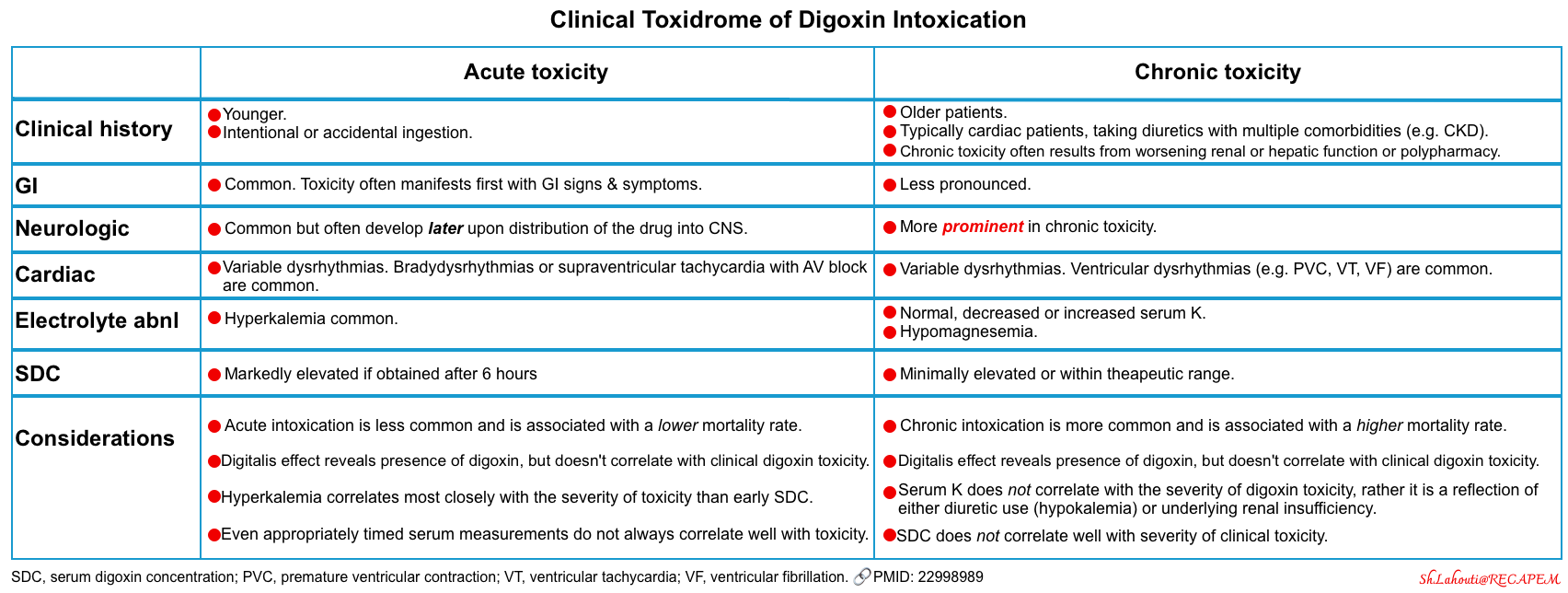
The timing and clinical presentation of acute and chronic digoxin toxicity differ significantly (table below).
- In acute intoxication, the patient may remain asymptomatic for several hours and then develop significant gastrointestinal symptoms. Neurologic manifestations are common and often develop later upon distribution of the drug into the central nervous system *. Hyperkalemia is more common and most closely correlates with the severity of clinical toxicity.
- Patients with chronic toxicity are brought to medical attention by family members who note a change in mental status or cognition. Gastrointestinal symptoms can occur but may be less pronounced. Neurologic manifestations may be prominent in chronic toxicity *.
- Serum K may be normal, decreased, or increased.
- Serum K does not correlate with the severity of digoxin toxicity, rather it is a reflection of either diuretic use (hypokalemia) or underlying renal insufficiency.
- Hypokalemia is more common and is more worrisome. It has been shown that hypokalemia, hypomagnesemia, and hypercalcemia tend to augment the toxicity of digoxin (even at therapeutic serum concentrations).
Differential diagnosis
Bradyarrhythmias (more on this, here)
- β-blocker or calcium channel blocker intoxication.
- Antiarrhythmics intoxication: Class IA antidysrhythmics (procainamide, quinidine), class IC antidysrhythmics (flecainide and encainide).
- Alpha-agonist intoxication (e.g. clonidine).
- Patients intoxicated with clonidine often have greater CNS depression, respiratory depression, and miosis.
- Organophosphate or carbamate insecticide poisoning.
- Toxidrome: Miosis & muscle spasm, sweating, lacrimation, salivation, urination, bronchorrhea, seizure/coma.
- Sick sinus syndrome, with its combination of supraventricular arrhythmias and cardiac conduction blocks.
- Hyperkalemia from any cause.
- Hypothermia
- Hypothyroidism
Delirium. Common causes of delirium and confusional states include:
- Medications *
- Sedative-hypnotics
- Antihistamines
- Anticholinergics: Tricyclic antidepressants (TCAs), atropine products, antiparkinsonian agents *
- Opioids
- Skeletal muscle relaxers (especially baclofen).
- Antipsychotics
- Lithium
- Metoclopramide
- Antiseizure medications (carbamazepine, phenytoin)
- Steroid
- Medication side effects (e.g. hyperammonemia from valproic acid *,*, confusion from quinolones, serotonin syndrome *)
- Polypharmacy
- Toxicologic
- Intoxication: Almost any substance (digoxin, toxic alcohols, lithium, carbon monoxide, sympathomimetic, sympatholytics, e.g. β-blockers, clonidine).
- Withdrawal from benzodiazepines, ethanol, opioids, and dexmedetomidine.
- Systemic organ failure, and metabolic
- Cardiovascular:
- Shock (hypoperfusion)
- Congestive encephalopathy *.
- PRES (Posterior Reversible Encephalopathy Syndrome).
- Infection: Sepsis (septic encephalopathy) *.
- Fever-related delirium.
- Hematologic: thrombocytosis, hypereosinophilia, leukemic blast cell crisis, polycythemia.
- Liver failure: acute, chronic
- Hepatic encephalopathy
- Accumulation of hepatically cleared medications
- Pulmonary disease, including hypercarbia and hypoxemia.
- Renal failure: acute, chronic
- Uremia
- Electrolyte disturbance especially ↑or ↓sodium, hypercalcemia.
- Accumulation of renally cleared medications e.g. digoxin.
- Endocrine disturbance:
- Thyroid storm, or myxedema coma.
- Adrenal crisis.
- Hypoglycemia, DKA, or hyperosmolar hyperglycemic state (HHS).
- Nutritional: Wernicke encephalopathy (thiamine deficiency), vitamin B12 deficiency, possibly folate and niacin deficiencies.
- Cardiovascular:
- Brain disorders
- CNS infections: encephalitis, meningitis, brain or epidural abscess.
- Autoimmune encephalitis.
- Ischemic stroke.
- Space occupying lesions: Intracranial hemorrhage, tumor.
- Traumatic brain injury.
- Epileptic seizures, especially nonconvulsive status epilepticus *.
- Psychiatric disorders
- Physical/Environmental
- Sleep deprivation
- Uncontrolled pain.
- Hyperthermia, hypothermia.
- Trauma: trauma with systemic inflammatory response syndrome, fat embolism.
Diagnosis
The diagnosis of digoxin toxicity is made clinically based on a combination of a history of exposure, suggestive clinical features, and/or ECG manifestations. Isolated elevated serum digoxin concentrations may confirm exposure but do not correlate consistently with clinical manifestations of toxicity.
Obtain history
- Determine the agent, amount, time of ingestion, and any coingestants when possible.
- Ask about symptoms suggesting an acute illness, such as gastroenteritis, that may have caused dehydration or acute renal insufficiency and contributed to the development of chronic toxicity.
- Inquire about GI, cardiac, and neurologic manifestations.
- Inquire also about symptoms that suggest hypoperfusion, such as confusion and abdominal pain, which may stem from mesenteric ischemia *.
- Obtain a thorough medication history to determine if any recent additions or dosing changes were made.
Physical exams
- Vital signs.
- Look for evidence of hypoperfusion and end-organ dysfunction.
- Pay particular attention to mentation and neurologic status, keeping in mind that such effects are often due to direct toxicity but may be secondary to cerebral hypoperfusion. Look for symptoms and signs suggestive of acute mesenteric ischemia, which is a rare complication.
Investigation
In the patient with suspected digoxin toxicity, the following studies should be obtained:
- Serial ECGs
- Fingerstick glucose, to rule out hypoglycemia as the cause of any alteration in mental status.
- Renal and liver function tests, electrolytes including Ca/Mg/Phos (if hypocalcemia is suspected check ionized calcium), and TSH if thyroid disease is suspected.
- Acetaminophen and salicylate levels, to rule out these common coingestants.
- Serum digoxin concentration (SDC).
- For an acute overdose, obtain a serum concentration measurement on presentation and approximately six hours after the ingestion; for chronic toxicity, obtain a concentration on presentation and interpret it in the context of the last administered dose (>6 hours for post-distribution level).
ECG
- Digitalis effects: Presence of the digitalis effect is useful to identify patients with exposure to cardiac glycosides but does not indicate toxicity.
- Dysrhythmias as explored above.
- Generally, almost any arrhythmia can occur, except atrial tachyarrhythmias with a rapid ventricular response (as these usually require intact conduction in the AVN).
- Paced ECG: In patients with a pacemaker in place, it might be difficult to diagnose digoxin intoxication as it may obscure the digitalis effect and also prevent the development of some arrhythmias *.
Serum digoxin level
- A quantitative serum digoxin level is readily measured in most hospital laboratories.
- Performance
- The serum digoxin level does not necessarily correlate with toxicity.
- Several reports have described asymptomatic patients with a “toxic” level, while others described patients with significant toxicity despite a serum digoxin level in the therapeutic range *.
- Nevertheless, an observational study of 171 digoxin poisonings reported a correlation between a higher mean serum digoxin concentration and 30-day mortality *.
- Following the administration of DSFab, serum immunoassays of digoxin are unreliable, as they measure both bound and unbound drugs.
- Fab treatment frequently causes an elevation in the serum digoxin concentration despite a free digoxin level approaching zero *.
- Elevated digoxin levels have been reported in the following conditions due to endogenous digoxin-like substances *:
- Newborns, and late-stage pregnancy (especially with pre-eclampsia).
- Patients with acromegaly.
- Subarachnoid hemorrhage.
- Renal failure, and liver disease.
- The digoxin immunoassay has cross-reactivity with other cardiac glycosides such as those found in various plant species.
- In patients with non-digoxin glycosides, digoxin level may be used as a qualitative assay (i.e. in an appropriate clinical context, a positive assay could be helpful to support the diagnosis of a non-digoxin cardiac glycoside intoxication).
- Digoxin level (quantitative assay) should not be used to calculate antidotal therapy with DSFab due to the following reasons:
- Incomplete cross-reactivity
- Lack of a correlation between the digoxin level and degree of toxicity.
- Digoxin level (quantitative assay) should not be used to calculate antidotal therapy with DSFab due to the following reasons:
- In patients with non-digoxin glycosides, digoxin level may be used as a qualitative assay (i.e. in an appropriate clinical context, a positive assay could be helpful to support the diagnosis of a non-digoxin cardiac glycoside intoxication).
- The serum digoxin level does not necessarily correlate with toxicity.
Indications for digoxin immunoassay
- When the clinical constellation is suggestive of cardiac glycoside intoxication.
- In patients with chronic digoxin use and the presence of any of the following *:
- Any signs or symptoms suggestive of digoxin poisoning.
- Acutely ill patients.
- Renal failure.
Timing of digoxin level
- Digoxin requires four hours after an IV dose or six hours after an oral dose to distribute into the tissues. Only post-distribution levels reflect the severity of intoxication and help calculate the dose of antiserum *.
- The serum digoxin level is likely to be falsely elevated if the sample is obtained soon after administration or ingestion because of the time required to equilibrate through the large volume of distribution.
- For acute intoxication, check a baseline digoxin level and then repeat another level six hours after the ingestion.
- A single digoxin level is adequate for chronic intoxication, provided that it is obtained >6 hours after the last oral dose.
Interpretation of the digoxin level in digoxin intoxication
- The therapeutic range for patients with heart failure is 0.5 to 0.9 ng/mL *, with a typical immunoassay reference range being 0.8 to 2.0 ng/mL.
- Digoxin level associated with severe toxicity:
- Acute intoxication: >10 ng/ml.
- Chronic intoxication: >4 ng/ml.
💡Knowledge of the digoxin level may help guide initial therapy, but the diagnosis and the decision to administer digoxin-Fab should not be based exclusively on the digoxin concentration, as clinical factors are more important.
General approach to management
- Assess and support airway, breathing, and circulation.
- Digoxin toxicity alone is not anticipated to cause significant mental status, airway, or breathing compromise.
- ⚠️If digoxin toxicity is considered, caution is advised in the use of succinylcholine as a paralyzing agent for intubation, given concerns related to hyperkalemia.
- Secure IV access.
- Initiate continuous cardiac monitoring.
- A 12-lead ECG.
- Fingerstick glucose, to rule out hypoglycemia as the cause of any alteration in mental status.
- Order serum digoxin, and labs.
- Renal and liver function tests, electrolytes including Ca/Mg/Phos (if hypocalcemia is suspected check ionized calcium), and TSH if thyroid disease is suspected.
- Hemodynamic assessment (e.g. bedside echo/ultrasound).
- Patients with chronic digoxin toxicity may require fluid.
- If indicated, IV fluid should be used carefully as many patients on digoxin have severe underlying heart disease.
- Assess for the indication of DSFab early upon suspicion and if indicated; order it promptly, as there is a substantial time delay between ordering DSFab and seeing a clinical improvement (generally at least ~2 hours).
GI decontamination
- GI decontamination with activated charcoal (1 g/kg PO), in an awake, alert, cooperative patient who presents within 1to2 h of ingestion is indicated.
- Insertion of a nasogastric tube in a non-intubated patient solely to administer activated charcoal is not recommended.
- Gastric lavage is not recommended.
- Asystole during lavage has been reported in a digoxin-toxic patient, presumably from vagal stimulation during the procedure, and no clinical benefit has been demonstrated.
Hemodynamic resuscitation
In intoxicated patients with cardiac glycosides and hemodynamically significant dysrhythmias, the treatment of choice is DSFab. The following treatments should be considered as a temporizing measure awaiting DSFab.
Bradycardia
- As a bridge to definitive treatment (digoxin-specific antibody fragments), the following measures should be considered to be taken:
- Atropine
- Atropine may work since patients with digoxin toxicity have excess vagal tone.
- Dosage: Start with 1 mg and repeat as needed, to a maximum total cumulative dose of 3 mg *.
- Temporary pacing (transcutaneous or transvenous) while awaiting digoxin-specific antibody fragments for symptomatic bradycardia that does not respond to atropine *.
- Hyperkalemia: If hyperkalemia is believed to be contributing to hemodynamic instability (e.g. causing AV block and bradycardia), then treatment should be started while awaiting DSFab. More on this, below.
- Atropine
Tachycardia
- Cardioversion
- ⚠️There is a relative contraindication to cardioversion in the setting of digitalis toxicity since digitalis sensitizes the heart to the electrical stimulus and, hence, cardioversion could trigger additional arrhythmias, most importantly ventricular fibrillation (VF).
- Supraventricular tachycardias
- Avoid cardioversion if possible for supraventricular tachycardias, given concern for inducing ventricular tachycardia *.
- Use the lowest energy that is likely to be successful, If urgent restoration of sinus rhythm is necessary for a hemodynamically unstable supraventricular arrhythmia.
- Rapid atrial pacing may be successful for certain arrhythmias (eg, atrial flutter) and is probably the safest method.
- Ventricular arrhythmias
- If cardioversion must be performed for life-threatening ventricular arrhythmia, prophylactic lidocaine (1 mg/kg up to a maximum dose of 100 mg IV push) should be given, and the lowest indicated energy levels (e.g. 20–100 J) used *,*.
- Cardioversion should be reserved only if VT is refractory to other treatments (e.g. lidocaine, Mg).
- When time permits, hypokalemia and hypomagnesemia should be corrected prior to cardioversion.
- Antiarrhythmics
- Lidocaine: Is the first-line agent for ventricular tachycardia *.
- It is given as 1- to 1.5-mg/kg IV bolus followed by continuous infusion at 1 to 4 mg/min in adults.
- Class IA antidysrhythmics (eg, quinidine, procainamide, disopyramide), and amiodarone are contraindicated in the setting of digoxin poisoning because they may induce or worsen AV nodal block by increasing vagal tone and decreasing His-Purkinje conduction by blocking fast Na channel opening.
- Lidocaine: Is the first-line agent for ventricular tachycardia *.
- Hypomagnesemia should be corrected if it is present. IV magnesium may counteract ventricular irritability seen with cardiac-glycoside toxicity. More on here.
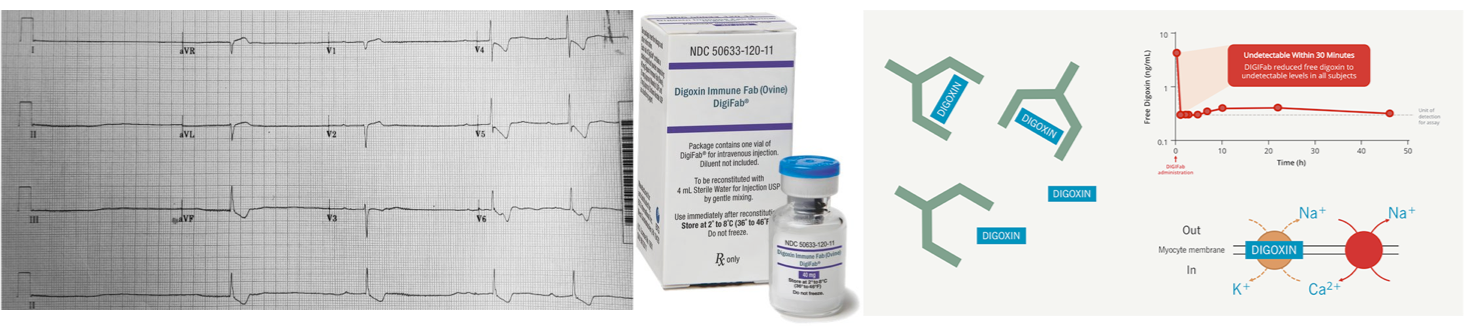
Digoxin-specific antibody fragments (DSFab)
Indications
- Any digoxin-related life-threatening dysrhythmias, regardless of serum digoxin level.
- Includes ventricular tachycardia, ventricular fibrillation, or progressive bradydysrhythmias such as atropine-resistant symptomatic sinus bradycardia or second or third-degree heart block.
- Potassium level >5 mEq/L in the setting of acute digoxin poisoning.
- Serum digoxin level ≥15 ng/mL at any time or ≥10-12 ng/mL 6 h post-ingestion, regardless of clinical effects *.
- Acute ingestion of 10 mg of digoxin in an adult ( 4 mg of digoxin in a child).
- Evidence of end-organ dysfunction from hypoperfusion (eg, renal failure, altered mental status)
- Chronic elevation of serum digoxin level that is associated with dysrhythmias, significant GI symptoms, or altered mental status.
- While altered mental status alone is not an indication for administration of digoxin-Fab in an otherwise hemodynamically stable person without arrhythmias, if the altered mental status is reflecting poor perfusion due to a bradyarrhythmia, digoxin-Fab is indicated.
- Moderate to severe gastrointestinal symptoms *.
Mechanism of action, and pharmacokinetic
- DSFab binds and neutralizes free digoxin molecules. As the level of free drugs falls, the resulting concentration gradient facilitates the dissociation of digoxin bound to Na/K-ATPase.
- The median time for initial response to Fab is ~20 minutes. However complete resolution of toxicity may take several hours (roughly 1.5-3 hours).
- Cardiac manifestations of toxicity usually subside within 30 minutes *.
- Hyperkalemia usually requires more time for redistribution and resolves within 2 to 6 hours.
- DSFab complex (DSFab bound to digoxin) is cleared by the kidney with an elimination half-life of ~15-20 hours (in patients with normal renal function) *.
- Dose adjustment is not required in patients with renal insufficiency.
- Since it is a large molecule, hemodialysis is not effective in removing it.
- Lab assay for serum digoxin level after administration of DSFab is useless.
- Because most lab assays measure bound and unbound digoxin, digoxin levels remain elevated for 1 week with values often > 100ng/mL after DSFab administration.
Adverse effects
- Hypokalemia (4%) *. It happens due to potassium shifting into cells.
- Heart failure or atrial fibrillation with rapid ventricular response (3%) may happen due to the removal of the digoxin effect.
- Allergic reactions may happen in ~1% of infusions *.
Rebound toxicity
- Refers to the reappearance of toxicity after an initial response to digoxin-specific antibody fragments.
- It may happen in ~2% of patients given a full neutralizing dose *.
- This can develop 12–24 hours after treatment, but up to 10 days later in patients with renal failure.
- Pathophysiology
- DSFab is generally excreted in the kidneys with a half-life of ~15-20 hours. Digoxin has a longer half-life (~40 hours).
- Therefore, it is possible that DSFab could be renally excreted, and subsequently, free digoxin levels could begin to rise again, leading to rebound toxicity.
- The binding of digoxin to DSFab is reversible, and dissociation occurs *.
- Patients with renal insufficiency are at risk of rebound toxicity since the renal clearance of the DSFab complex is delayed, and this complex may dissociate several days later.
- DSFab is generally excreted in the kidneys with a half-life of ~15-20 hours. Digoxin has a longer half-life (~40 hours).
- Serum digoxin concentration is of no use in diagnosis because it measures the digoxin in the complexes with antibody fragments as well as unbound digoxin.
- The concentration therefore rises many-fold after DSFab is given (even in the absence of rebound toxicity) *.
- Treatment: Re-administration of DSFab.
Dosage
- Two brands are available, Digibind and DigiFab, which seem interchangeable.
- In acute poisoning, each vial of either product reverses approximately 0.5 mg of ingested digoxin.
A full neutralizing dose of digoxin-Fab is based on an estimation of either the dose ingested or a steady-state serum digoxin level.
- When neither the serum level nor the amount of ingestion is known (empiric administration). The dosage depends on the severity of the toxicity.
- Patients with cardiac arrest or peri-arrest
- 10 vials in adults or 5 vials in children; slow IV push. Reevaluate clinically in 30 minutes.
- The initial dose can be repeated if there is inadequate clinical response (although at this time, it is reasonable to reconsider the diagnosis of digoxin toxicity). Watch for volume overload in small children.
- 10 vials in adults or 5 vials in children; slow IV push. Reevaluate clinically in 30 minutes.
- Patient with relative hemodynamic stability
- Patients with cardiac arrest or peri-arrest
- The amount of digoxin ingested is known but the concentration is unknown
- Number of vials= Ingested digoxin (mg) x1.6
- Steady-state digoxin concentration is known
- Number of vials= Serum concentration (ng/mL) × weight (kg)/100
- MDCalc: Digibind Dosing for Digoxin Poisoning: MDClac can help to determine the number of vials of DSFab.
Administration
- Rate of IV infusion
- Patients with cardiac arrest or peri-arrest: Slow IV Push.
- All other patients: Fab fragments should be given over 30 minutes.
- Partial reversal vs. full neutralizing dose.
- Partial reversal dosage (half the recommended dosage) can be considered for patients with chronic toxicity who are hemodynamically stable without life-threatening dysrhythmias (eg, AV nodal blockade present on ECG but the patient maintains normal blood pressure and clear mentation).
- This strategy can be employed to avoid unmasking the condition for which the patient is taking digoxin.
- Giving a full dose can elicit acute decompensated heart failure or atrial fibrillation with rapid ventricular response.
- Treatment can be started with 3 vials and followed clinically to determine whether additional treatment is warranted *.
- This strategy can be employed to avoid unmasking the condition for which the patient is taking digoxin.
- Partial reversal dosage (half the recommended dosage) can be considered for patients with chronic toxicity who are hemodynamically stable without life-threatening dysrhythmias (eg, AV nodal blockade present on ECG but the patient maintains normal blood pressure and clear mentation).
Electrolyte therapy
Hyperkalemia
- In acute digoxin intoxication, hyperkalemia is an indicator of severe toxicity.
- The treatment of choice in this setting is DSFab.
- Traditional treatment of hyperkalemia may be started while awaiting DSFab, if hyperkalemia is believed to be contributing to hemodynamic instability (e.g. bradycardia).
- Principle: Do not aggressively treat hyperkalemia.
- In digoxin-induced hyperkalemia, mild hyperkalemia may be protective against digoxin toxicity. It may help to drive potassium into cardiomyocytes via Na/K-ATPase, and will also occupy more K-binding sites on the Na/K-ATPase pump thereby fewer binding sites will be available for digoxin.
- DSFab will cause a potassium shift into cells, so aggressive treatment of hyperkalemia plus DSFab could cause an overshoot hypokalemia.
- Treatment of hyperkalemia (if indicated) includes
- IV insulin and glucose.
- Potassium-wasting diuretics plus crystalloid (to facilitate renal potassium excretion).
- IV Calcium in digoxin–induced hyperkalemia is controversial.
- Older literature indicated an increased incidence of ventricular arrhythmias and a higher mortality when calcium was administered to digoxin-toxic patients. Theoretically, IV calcium can have the additive or synergistic actions of calcium and CAS on the heart (because intracellular hypercalcemia is already present), resulting in dysrhythmias, cardiac dysfunction (e.g. hyper contractility, so-called stone heart hypocontractility), and cardiac arrest.
- However, data from an animal model and retrospective review of a limited number of human patients with digoxin toxicity who received IV calcium found no increase in ventricular arrhythmias or mortality *,*,*.
- Given that hyperkalemia is not typically the cause of death in digoxin toxicity, calcium salts are best avoided in cases of recognized digoxin toxicity.
- Principle: Do not aggressively treat hyperkalemia.
Hypokalemia
- Hypokalemia is common among patients with chronic digoxin intoxication. This is secondary to contributory factors such as long-term diuretic use to treat congestive heart failure.
- Hypokalemia can exacerbate/worsen digoxin intoxication and should be corrected by potassium replacement. More on this, here.
- If DSFab is administered, this may cause potassium to enter the cells, thereby exacerbating the hypokalemia.
- DSFab administration generally should be withheld until the hypokalemia is corrected *.
Hypomagnesemia
- Magnesium is a cofactor in the Na/K exchange channel, and hypomagnesemia can exacerbate/worsen digoxin intoxication.
- Hypomagnesemia should be corrected.
- The theoretical benefits of magnesium therapy in the setting of hypomagnesemia include
- Blockade of the transient inward calcium current.
- Antagonism of calcium at intracellular binding sites.
- Decreased digoxin-related ventricular irritability.
- Blockade of potassium escape from digoxin-poisoned cells.
- More on Treatment: Cardiac Mg Protocol
Non-digoxin CAS (e.g. plants like foxglove)
- Digoxin levels in these situations are not quantitatively meaningful. Therefore, DSFab must be dosed empirically based on clinical severity.
- DSFab may have a lower affinity for non-digoxin cardiac glycosides. Therefore, larger doses may be needed than for digoxin intoxication.
- For patients with critical cardiac dysfunction, 10-20 vials may be used *.
Cardiac arrest
- Perform CPR with current advanced cardiac life support protocols (prolonged CPR may be appropriate *).
- Resuscitation efforts should be continued for at least 30 minutes after giving digoxin-specific antibody fragments *.
- Digoxin-specific antibody fragments: IV bolus (10 vials if the amount ingested is unknown).
Rapid review

Media
Going further
- ToxCard: Digoxin Toxicity (emDocs)
- Tox and Hound – To Bind or Not To Bind(EMCrit)
- Cardiac glycoside poisoning (including digoxin): IBCC
- Digoxin toxicity (CORE EM)
References
1. Brubacher JR, Lachmanen D, Ravikumar PR, Hoffman RS. Efficacy of digoxin-specific Fab fragments (Digibind) in the treatment of toad venom poisoning. Toxicon. 1999 Jun;37(6):931-42. doi: 10.1016/s0041-0101(98)00224-4. PMID: 10340832.
2. Botelho AFM, Pierezan F, Soto-Blanco B, Melo MM. A review of cardiac glycosides: Structure, toxicokinetics, clinical signs, diagnosis and antineoplastic potential. Toxicon. 2019 Feb;158:63-68. doi: 10.1016/j.toxicon.2018.11.429. Epub 2018 Dec 4. PMID: 30529380.
3.Ziff OJ, Kotecha D. Digoxin: The good and the bad. Trends Cardiovasc Med. 2016 Oct;26(7):585-95. doi: 10.1016/j.tcm.2016.03.011. Epub 2016 Mar 31. PMID: 27156593.
4.Desai MY, Watanabe MA, Laddu AA, Hauptman PJ. Pharmacologic modulation of parasympathetic activity in heart failure. Heart Fail Rev. 2011 Mar;16(2):179-93. doi: 10.1007/s10741-010-9195-1. PMID: 20924667
5. Sticherling C, Oral H, Horrocks J, Chough SP, Baker RL, Kim MH, Wasmer K, Pelosi F, Knight BP, Michaud GF, Strickberger SA, Morady F. Effects of digoxin on acute, atrial fibrillation-induced changes in atrial refractoriness. Circulation. 2000 Nov 14;102(20):2503-8. doi: 10.1161/01.cir.102.20.2503. PMID: 11076824.
6. Pace DG, Gillis RA. Neuroexcitatory effects of digoxin in the cat. J Pharmacol Exp Ther. 1976 Dec;199(3):583-600. PMID: 994017
7. Worthley LI, Holt AW. Digoxin in the critically ill patient. Crit Care Resusc. 1999 Sep;1(3):252-64. PMID: 16603014.
8. Ahmed A, Pitt B, Rahimtoola SH, Waagstein F, White M, Love TE, Braunwald E. Effects of digoxin at low serum concentrations on mortality and hospitalization in heart failure: a propensity-matched study of the DIG trial. Int J Cardiol. 2008 Jan 11;123(2):138-46. doi: 10.1016/j.ijcard.2006.12.001. Epub 2007 Mar 23. PMID: 17382417; PMCID: PMC2474767
9. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013 Oct 15;62(16):e147-239. doi: 10.1016/j.jacc.2013.05.019. Epub 2013 Jun 5. PMID: 23747642.
10. Goldberger ZD, Goldberger AL. Therapeutic ranges of serum digoxin concentrations in patients with heart failure. Am J Cardiol. 2012 Jun 15;109(12):1818-21. doi: 10.1016/j.amjcard.2012.02.028. Epub 2012 Apr 11. PMID: 22502901; PMCID: PMC3646412.
11. Ehle M, Patel C, Giugliano RP. Digoxin: clinical highlights: a review of digoxin and its use in contemporary medicine. Crit Pathw Cardiol. 2011 Jun;10(2):93-8. doi: 10.1097/HPC.0b013e318221e7dd. PMID: 21988950.
12. Eichhorn EJ, Gheorghiade M. Digoxin. Prog Cardiovasc Dis. 2002 Jan-Feb;44(4):251-66. doi: 10.1053/pcad.2002.31591. PMID: 12007081.
13. Ambrosy AP, Pang PS, Gheorghiade M. Digoxin for Worsening Chronic Heart Failure: Underutilized and Underrated. JACC Heart Fail. 2016 May;4(5):365-7. doi: 10.1016/j.jchf.2016.03.005. PMID: 27126283.
14. Pincus M. Management of digoxin toxicity. Aust Prescr. 2016 Feb;39(1):18-20. doi: 10.18773/austprescr.2016.006. Epub 2016 Feb 1. PMID: 27041802; PMCID: PMC4816869.
15. Ma G, Brady WJ, Pollack M, Chan TC. Electrocardiographic manifestations: digitalis toxicity. J Emerg Med. 2001 Feb;20(2):145-52. doi: 10.1016/s0736-4679(00)00312-7. PMID: 11207409.
16. Almarzuqi A, Kimber S, Quadros K, Senaratne J. Bidirectional Ventricular Tachycardia: Challenges and Solutions. Vasc Health Risk Manag. 2022 Jun 7;18:397-406. doi: 10.2147/VHRM.S274857. PMID: 35698640; PMCID: PMC9188370.
17. Manne JRR. Regularized Atrial Fibrillation. Am J Med. 2018 Sep;131(9):e361-e363. doi: 10.1016/j.amjmed.2018.04.010. Epub 2018 May 1. PMID: 29723516.
Nelson L, Goldfrank LR. Goldfrank’s Toxicologic emergencies. 11th ed. New York: McGraw-Hill Medical; 2019.
18. Kanji S, MacLean RD. Cardiac glycoside toxicity: more than 200 years and counting. Crit Care Clin. 2012 Oct;28(4):527-35. doi: 10.1016/j.ccc.2012.07.005. Epub 2012 Aug 30. PMID: 22998989.
19. Francis, J. Drug-Induced Delirium. CNS Drugs 5, 103–114 (1996). https://doi.org/10.2165/00023210-199605020-00003.
20. Rudolph JL, Salow MJ, Angelini MC, McGlinchey RE. The anticholinergic risk scale and anticholinergic adverse effects in older persons. Arch Intern Med. 2008 Mar 10;168(5):508-13. doi: 10.1001/archinternmed.2007.106. PMID: 18332297.
21. Walker V. Severe hyperammonaemia in adults not explained by liver disease. Ann Clin Biochem. 2012 May;49(Pt 3):214-28. doi: 10.1258/acb.2011.011206. Epub 2012 Feb 20. PMID: 22349554.
22. Zhao L, Walline JH, Gao Y, Lu X, Yu S, Ge Z, Zhu H, Li Y. Prognostic Role of Ammonia in Critical Care Patients Without Known Hepatic Disease. Front Med (Lausanne). 2020 Oct 22;7:589825. doi: 10.3389/fmed.2020.589825. PMID: 33195354; PMCID: PMC7642587.
23. Beaubien-Souligny W, Cavayas YA, Denault A, Lamarche Y. First step toward uncovering perioperative congestive encephalopathy. J Thorac Cardiovasc Surg. 2020 Jul 2:S0022-5223(20)31087-4. doi: 10.1016/j.jtcvs.2020.02.146. Epub ahead of print. PMID: 32624312.
24. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012 Oct;8(10):557-66. doi: 10.1038/nrneurol.2012.183. Epub 2012 Sep 18. PMID: 22986430.
25. Veran O, Kahane P, Thomas P, Hamelin S, Sabourdy C, Vercueil L. De novo epileptic confusion in the elderly: a 1-year prospective study. Epilepsia. 2010 Jun;51(6):1030-5. doi: 10.1111/j.1528-1167.2009.02410.x. Epub 2009 Dec 1. PMID: 20002146.
26. Bridwell RE, Baker KA, Hoyte CO, Ng PC. Digoxin Toxicity in a Patient with Pacemaker: A Case Report. Cureus. 2019 Nov 2;11(11):e6056. doi: 10.7759/cureus.6056. PMID: 31819839; PMCID: PMC6886726.
27. Sonnenblick M, Abraham AS, Meshulam Z, Eylath U. Correlation between manifestations of digoxin toxicity and serum digoxin, calcium, potassium, and magnesium concentrations and arterial pH. Br Med J (Clin Res Ed). 1983 Apr 2;286(6371):1089-91. doi: 10.1136/bmj.286.6371.1089. PMID: 6404339; PMCID: PMC1547496.
28. Supervía Caparrós A, Salgado García E, Calpe Perarnau X, Galicia Paredes M, García Gibert L, Córdoba Ruiz F, Clemente Rodríguez C, Nogué Xarau S. Immediate and 30 days mortality in digoxin poisoning cases attended in the Hospital Emergency Services of Catalonia, Spain. Emergencias. 2019 Feb;31(1):39-42. English, Spanish. PMID: 30656872.
29. Flanagan RJ, Jones AL. Fab antibody fragments: some applications in clinical toxicology. Drug Saf. 2004;27(14):1115-33. doi: 10.2165/00002018-200427140-00004. PMID: 15554746.
30. Dasgupta A. Endogenous and exogenous digoxin-like immunoreactive substances: impact on therapeutic drug monitoring of digoxin. Am J Clin Pathol. 2002 Jul;118(1):132-40. doi: 10.1309/3VNP-TWFQ-HT9A-1QH8. PMID: 12109847.
31. Lewis RP. Clinical use of serum digoxin concentrations. Am J Cardiol. 1992 Jun 4;69(18):97G-106G; discussion 106G-107G. doi: 10.1016/0002-9149(92)91258-6. PMID: 1626495.
32. Roberts DM, Gallapatthy G, Dunuwille A, Chan BS. Pharmacological treatment of cardiac glycoside poisoning. Br J Clin Pharmacol. 2016 Mar;81(3):488-95. doi: 10.1111/bcp.12814. Epub 2015 Dec 15. PMID: 26505271; PMCID: PMC4767196.
33. Chen JY, Liu PY, Chen JH, Lin LJ. Safety of transvenous temporary cardiac pacing in patients with accidental digoxin overdose and symptomatic bradycardia. Cardiology. 2004;102(3):152-5. doi: 10.1159/000080483. Epub 2004 Aug 27. PMID: 15334025.
34. Sarubbi B, Ducceschi V, D’Andrea A, Liccardo B, Santangelo L, Iacono A. Atrial fibrillation: what are the effects of drug therapy on the effectiveness and complications of electrical cardioversion? Can J Cardiol. 1998 Oct;14(10):1267-73. PMID: 9852940.
35. Kelly RA, Smith TW. Recognition and management of digitalis toxicity. Am J Cardiol. 1992 Jun 4;69(18):108G-118G; disc. 118G-119G. doi: 10.1016/0002-9149(92)91259-7. PMID: 1626485.
36. Bhatia SJ. Digitalis toxicity–turning over a new leaf? West J Med. 1986 Jul;145(1):74-82. PMID: 3529634; PMCID: PMC1306817.
37. Bauman JL, Didomenico RJ, Galanter WL. Mechanisms, manifestations, and management of digoxin toxicity in the modern era. Am J Cardiovasc Drugs. 2006;6(2):77-86. doi: 10.2165/00129784-200606020-00002. PMID: 16555861
38. Hickey AR, Wenger TL, Carpenter VP, Tilson HH, Hlatky MA, Furberg CD, Kirkpatrick CH, Strauss HC, Smith TW. Digoxin Immune Fab therapy in the management of digitalis intoxication: safety and efficacy results of an observational surveillance study. J Am Coll Cardiol. 1991 Mar 1;17(3):590-8. doi: 10.1016/s0735-1097(10)80170-6. PMID: 1993775.
39. Ujhelyi MR, Robert S. Pharmacokinetic aspects of digoxin-specific Fab therapy in the management of digitalis toxicity. Clin Pharmacokinet. 1995 Jun;28(6):483-93. doi: 10.2165/00003088-199528060-00006. PMID: 7656506
40. Chan BS, Isbister GK, Chiew A, Isoardi K, Buckley NA. Clinical experience with titrating doses of digoxin antibodies in acute digoxin poisoning. (ATOM-6). Clin Toxicol (Phila). 2022 Apr;60(4):433-439. doi: 10.1080/15563650.2021.1968422. Epub 2021 Aug 23. PMID: 34424803.
41. Lucyk S. Calculated decisions: DigiFab® (Digibind®) Dosing for Digoxin Poisoning. Emerg Med Pract. 2020 Sep 15;22(Suppl 9):CD1-CD3. PMID: 33476509.
42. Hack JB, Woody JH, Lewis DE, Brewer K, Meggs WJ. The effect of calcium chloride in treating hyperkalemia due to acute digoxin toxicity in a porcine model. J Toxicol Clin Toxicol. 2004;42(4):337-42. doi: 10.1081/clt-120039538. PMID: 15461240.
43. Levine M, Nikkanen H, Pallin DJ. The effects of intravenous calcium in patients with digoxin toxicity. J Emerg Med. 2011 Jan;40(1):41-6. doi: 10.1016/j.jemermed.2008.09.027. Epub 2009 Feb 6. PMID: 19201134.
44. Van Deusen SK, Birkhahn RH, Gaeta TJ. Treatment of hyperkalemia in a patient with unrecognized digitalis toxicity. J Toxicol Clin Toxicol. 2003;41(4):373-6. doi: 10.1081/clt-120022006. PMID: 12870880.
45. Patel SH, Reynolds JC, Judge BS. Successful treatment of prolonged digoxin-induced cardiac arrest with mechanical chest compressions and digoxin-specific antibody fragments. Resuscitation. 2017 Jun;115:e7-e8. doi: 10.1016/j.resuscitation.2017.04.001. Epub 2017 Apr 4. PMID: 28385641
Add comment