

15 April 2024 by Shahriar Lahouti.
CONTENTS
- Preface
- Hypokinetic Movement Disorders
- Hyperkinetic Movement Disorders
- Appendix
- References
Preface
Most movement disorders are slowly progressive and neurodegenerative, however, there are many acute and emergent medical conditions in which a movement disorder becomes a predominant (or presenting) feature.
An emergent movement disorder has been defined as any neurological disorder evolving acutely or subacutely, in which a primary movement disorder dominates the clinical presentation, and in which failure to diagnose and manage the patient accurately may result in significant morbidity or even mortality.
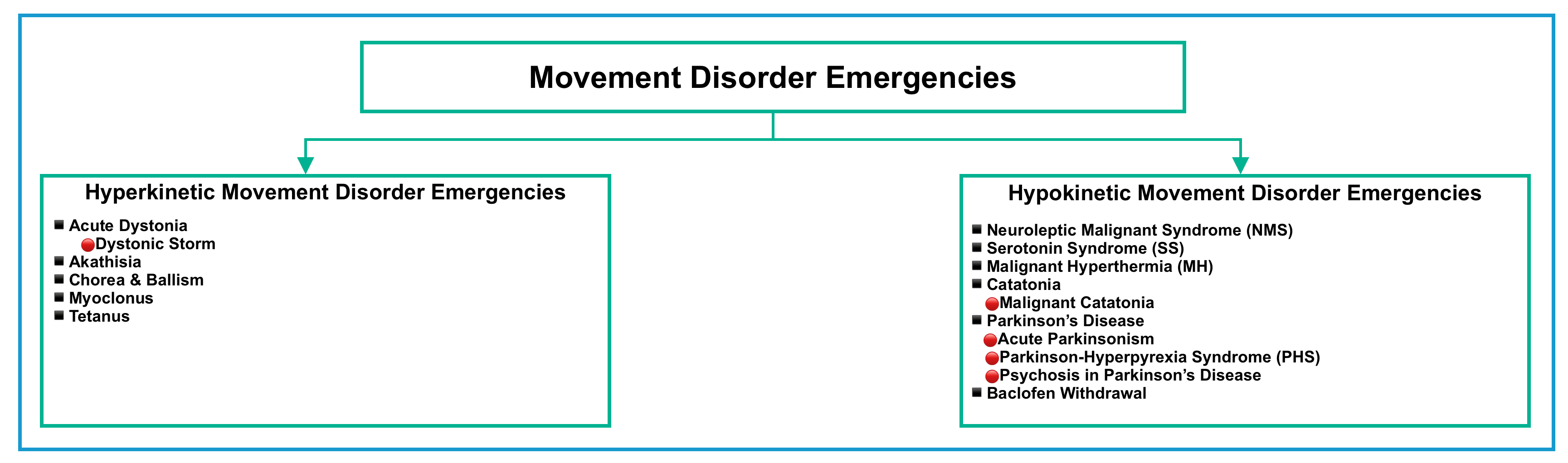
- Generally, movement disorders are broadly classified as either hypokinetic (decreased movement), or hyperkinetic (increased movement). This post discusses well-reported acute syndromes in emergency neurology (figure below).
- Note that some syndromes have features of both hypo-and-hyperkinetic movement disorders; for example, Parkinson’s disease presents with both hyperkinetic (tremor) and hypokinetic (bradykinesia) symptoms but is generally categorized as a hypokinetic movement disorder based on its predominant semiology.
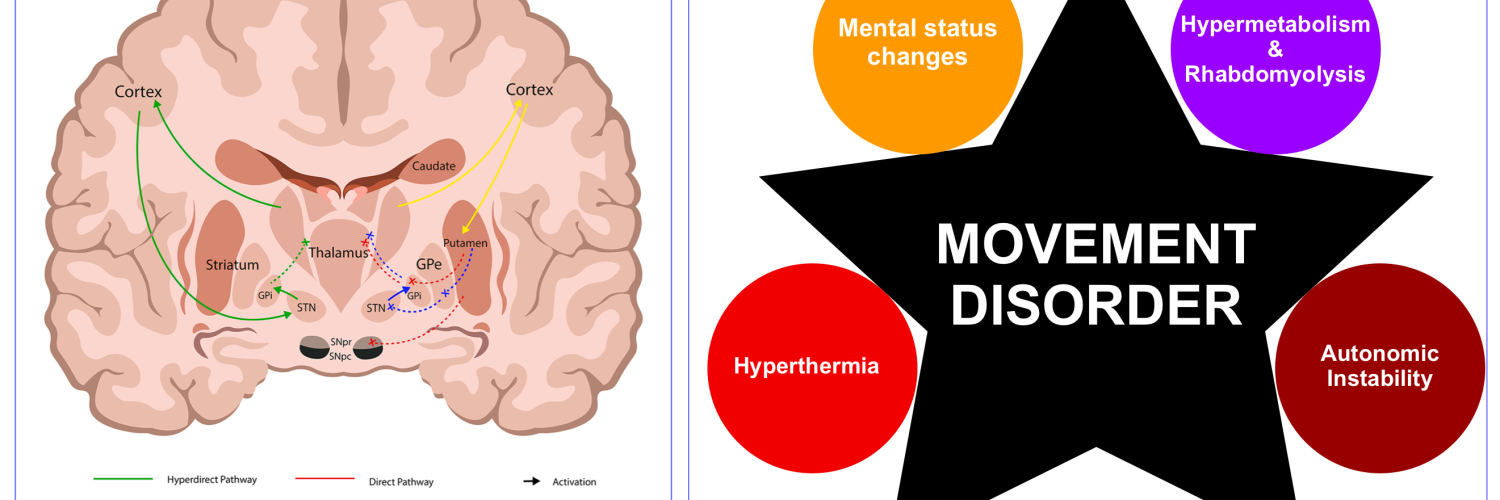
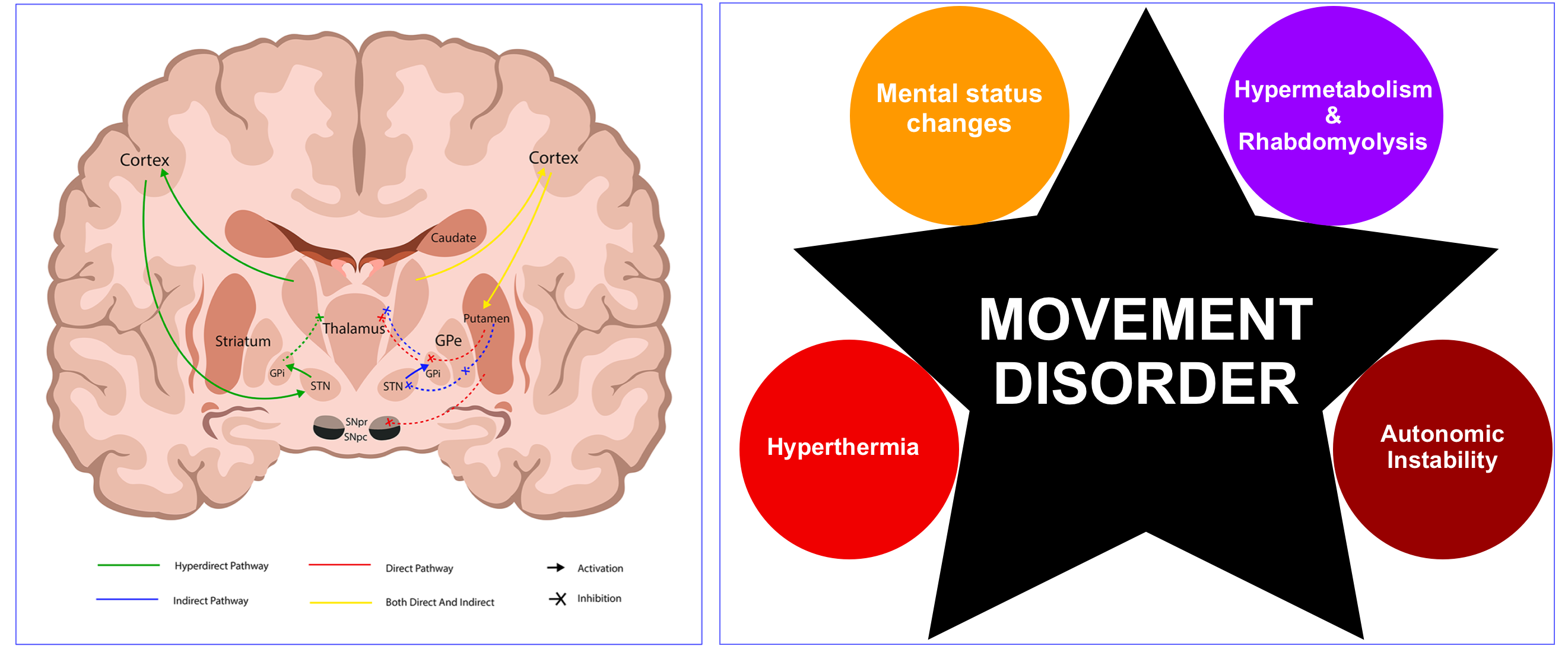
- The pathophysiology of many movement disorders (other than a few exceptions), involves diseases or drugs that impair neurotransmission in the basal ganglia.
- For example, most common hypokinetic disorders are caused by pathology affecting the dopaminergic projection from the substantia nigra of the midbrain to the putamen and caudate such as Parkinson’s disease or drugs that are antagonists at dopamine receptors, e.g. neuroleptic drugs.
- The diagnosis of a movement disorder is primarily made clinically *.
- For better diagnosis and management, the symptoms of movement disorders (e.g. rigidity) need to be put into perspective with other clinical features (such as delirium, dysautonomia, and hypermetabolic states).
| Hypokinetic Movement Disorders |
Hypokinetic disorders are characterized by rigidity (an increase in muscle tone that is equal in flexion and extension and throughout the range of movement), akinesia (reduced spontaneous movement), and bradykinesia (slowness of movement).
- Conventionally, motor weakness (paresis) and motor incoordination (ataxia) are not included, since their anatomy, diagnostic workup, and clinical management differ from hypokinetic and hyperkinetic disorders.
Neuroleptic malignant syndrome
▪️General
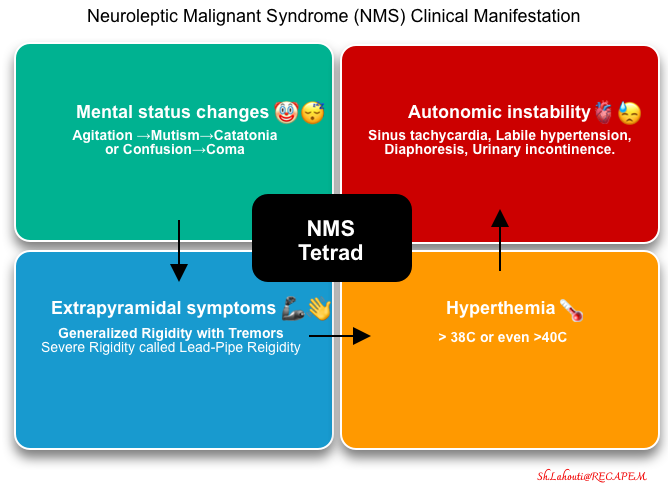
- Neuroleptic malignant syndrome (NMS) is a potentially lethal syndrome linked to exposure to dopamine antagonists or abrupt withdrawal of dopamine agonists *.
- The syndrome typically constitutes a clinical tetrad of altered mental status, hyperthermia, rigidity, and dysautonomia (figure below) *.
- However, the clinical presentation of NMS is heterogeneous, making diagnosis difficult, especially in the early phase.
- In clinical practice, NMS is not a clear and easily distinguishable syndrome *.
- NMS is a diagnosis of exclusion, as the differential diagnosis of the symptom complex is broad.
- NMS always needs to be kept in mind after administering a high-potency antipsychotic when developing signs of rigidity regardless of autonomic instability.
- Treatment of NMS includes withdrawal of the offending dopamine antagonist (or reinstatement of the dopamine agonist).
- In mild to moderate NMS, symptomatic therapy might be sufficient.
- Specific therapy for neuroleptic malignant syndrome ( Dantrolene, Bromocriptine, Electroconvulsive therapy) should be applied in severe cases *.
▪️Pathogenesis
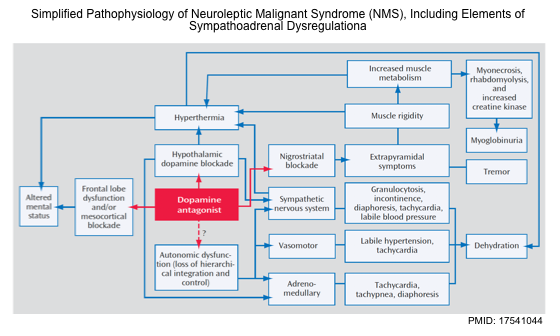
- The pathophysiology of NMS is likely complex (figure below), involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems culminating in an end-stage hypermetabolic syndrome *.
- The precise pathophysiological mechanisms of NMS are unproven, however, since NMS is associated with a certain class of medication (i.e. antidopaminergic agents), dopamine pathway deficiency is central to most theories of its pathogenesis *. The current theories are mentioned here.
- 🧠Central alteration of dopaminergic pathways
- Central dopamine receptor blockade in the hypothalamus may cause hyperthermia and other signs of dysautonomia.
- Dopamine deficiency in the striatal dopaminergic pathway within basal ganglia may cause Parkinsonian-type symptoms such as rigidity and tremor *.
- Dopamine alterations within the reticular activating system may cause altered mental status seen in patients with NMS *.
- Direct toxicity on the peripheral muscle system
- Dopamine deficiency may increase the release of calcium from the sarcoplasmic reticulum of muscle cells which could lead to rigidity, muscle breakdown, and hyperthermia seen in NMS *.
- Sympathoadrenal hyperactivity
- Dopamine plays a modulatory effect on efferent sympathetic tone.
- Dopamine antagonists may cause sympathoadrenal hyperactivity, resulting from the removal of tonic inhibition within the sympathetic nervous system *. This can explain the autonomic symptoms in NMS and the elevation in the urine and plasma catecholamine levels in patients with NMS.
- Sympathetic activation can generate heat through brown fat and skeletal muscle uncoupling proteins like amphetamines.
- Activation of the sympathetic nervous system may also impair heat dissipation through cutaneous vasoconstriction.
- 🧠Central alteration of dopaminergic pathways
▪️Etiology
- Antidopaminergic medications
- Neuroleptics (i.e. antipsychotic agents) see table in appendix 1, 2.
- Virtually all antipsychotics, including typical (e.g. haloperidol, chlorpromazine, thioridazine) and atypical antipsychotics (e.g. olanzapine, clozapine, quetiapine, aripiprazole), exhibit the potential to precipitate NMS.
- However, first-generation (typical) antipsychotics have a greater risk than “atypical” antipsychotics *.
- 👉Clozapine and quetiapine have the lowest risk.
- Non-neuroleptics
- Antiemetics with antidopaminergic activity: Prochlorperazine, promethazine, metoclopramide, domperidone, droperidol.
- Neuroleptics (i.e. antipsychotic agents) see table in appendix 1, 2.
- Withdrawal of dopaminergic antiparkinsonian medications
- Levodopa
- Dopamine receptor agonists: Bromocriptine, pramipexole, ropinirole, rotigotine, apomorphine.
- Amantadine
- Tolcapone (COMT inhibitors)
▪️Risk factors
- Previous episodes of NMS or catatonia * or a family history of NMS *.
- Dehydration, prolonged heat exposure *.
- Delirium, preexisting organic brain damage, alcohol, and drug addiction *.
- Neuroleptic therapy
- First-generation (typical) antipsychotics have a greater risk than “atypical” antipsychotics *.
- Parenteral antipsychotics *, higher doses of antipsychotics, or rapid titration of the desired therapeutic serum levels.
- Oily long-acting depot forms of neuroleptics (such as haloperidol decanoate)
- Multiple antipsychotic agents, co-medication with lithium *, and selective serotonin reuptake inhibitors (SSRIs)*.
- Underlying dopamine deficiency
- Senile dementia of Lewy-type, and Parkinson’s disease.
- Abrupt discontinuation of dopamine agonists during therapy of Parkinson’s disease.
- Other antidopaminergic medications
▪️Clinical features
- Chronocity 🕰
- It most often occurs within ~2 weeks after the start of antipsychotic therapy or after a dosage adjustment, however, it can occur any time during the use of antipsychotic drugs *.
- The antipsychotic serum concentration is usually within the therapeutic range.
- Symptoms usually develop gradually over 1-2 days, but timing is extremely variable.
- NMS usually resolves over ~2 weeks, but depot preparations or complications can prolong the course.
- Clinical presentation
- Mental status changes and extrapyramidal symptoms often precede hyperthermia and autonomic instability. Initially, many patients will only have mental status changes and extrapyramidal features, which may render early diagnosis challenging.
- Altered mental status 🤡😴
- Agitation may occur initially, with later evolution into mutism, catatonia, and coma.
- These symptoms may be obscured in patients who are intubated and sedated, or in patients with poorly controlled schizophrenia (in whom these symptoms may blend into the symptoms of schizophrenia).
- Extrapyramidal symptoms 🦾, 👋
- Hyperthermia 🥵
- Not all patients have hyperthermia:
- Temp > 38C is present in almost ~87% of patients *.
- Temp > 40C is present in almost ~40% of patients (so frank hyperthermia is often absent).
- ⚠️Delayed onset of hyperthermia may promote diagnostic confusion.
- Similar to other forms of hyperthermia, this is usually not associated with chills, nor responsive to antipyretic therapy.
- Not all patients have hyperthermia:
- Dysautonomia (autonomic instability with sympathetic activation)
- Altered mental status 🤡😴
- Mental status changes and extrapyramidal symptoms often precede hyperthermia and autonomic instability. Initially, many patients will only have mental status changes and extrapyramidal features, which may render early diagnosis challenging.
- Associated symptoms may include myoclonus, seizures, ataxia, hyporeflexia, and extensor plantar response *.
🔴The clinical features of NMS associated with atypical antipsychotics differ somewhat from the classic presentation induced by the typical antipsychotics. Fever and sympathetic liability are less common in NMS induced by atypical antipsychotics *.
▪️Complications
- Renal failure due to rhabdomyolysis, and electrolyte imbalance.
- Venous thromboembolic events and pulmonary embolism (immobility).
- Takotsubo cardiomyopathy (autonomic instability).
- Seizures.
- Aspiration, pneumonia, and respiratory failure (dysphagia and hypersalivation caused by NMS).
- Disseminated intravascular coagulation (DIC) and multiorgan failure (hyperthermia).
▪️Laboratory abnormality
- Creatinine kinase is often >1,000 U/L (~70% of patients with NMS) *.
- CPK is used as an indicator of rigidity severity.
- Leukocytosis (as high as 40,000), left shift, and ⬆️neutrophil/lymphocyte ratio (~38% of patients *).
- ⬆️CRP is often present, suggesting that NMS may involve widespread inflammation.
▪️Differential diagnosis: Differential diagnosis is of prime importance because NMS is a diagnosis of exclusion *.
- Infectious
- Meningitis or encephalitis, brain abscess, tetanus.
- Sepsis
- Toxic or pharmacological
- Malignant hyperthermia (inhalational anesthetics, succinylcholine)
- Serotonin syndrome
- Clinical features more indicative of serotonin syndrome include a more rapid onset (2 to 24 hours), shivering, hyperreflexia, myoclonus, nausea, vomiting, diarrhea, and less intense muscle rigidity and fever.
- Sympathomimetics intoxication (cocaine, ecstasy, or methamphetamines).
- Anticholinergic delirium
- Salicylate poisoning
- Lithium intoxication.
- Withdrawal from dopamine agonists, baclofen, sedative-hypnotics, and alcohol.
- Psychiatric or neurological
- Nonconvulsive status epilepticus.
- Structural lesions, particularly involving the midbrain, and basal ganglia.
- Acute Parkinsonism.
- Benign extrapyramidal side effects of a medication (e.g. isolated dystonic reaction).
- Malignant catatonia
- NMS is one subtype of malignant catatonia that is triggered by neuroleptics. Thus, neuroleptic malignant syndrome is often indistinguishable from other forms of malignant catatonia. Other potential triggers of catatonia should therefore be considered. Features that may suggest an additional or alternative cause of catatonia may include:
- Behavioral prodrome for weeks involving psychosis, agitation, or apathy *.
- More positive motor symptoms (e.g. waxy flexibility, stereotyped repetitive movements, automatisms).
- NMS is one subtype of malignant catatonia that is triggered by neuroleptics. Thus, neuroleptic malignant syndrome is often indistinguishable from other forms of malignant catatonia. Other potential triggers of catatonia should therefore be considered. Features that may suggest an additional or alternative cause of catatonia may include:
- Agitated delirium
- Endocrine
- Thyrotoxicosis (may cause tremor, but extrapyramidal signs are absent), pheochromocytoma.
- Environmental heatstroke
- In heat stroke, in addition to a history of exertion or exposure to high ambient temperature, skin is dry and muscle flaccidity is commonly observed *.
- Importantly, neuroleptic medications can predispose patients to hyperthermia, making them prone to heat stroke, especially if contributing factors such as hot weather, dehydration, excessive exercise, or agitation are present
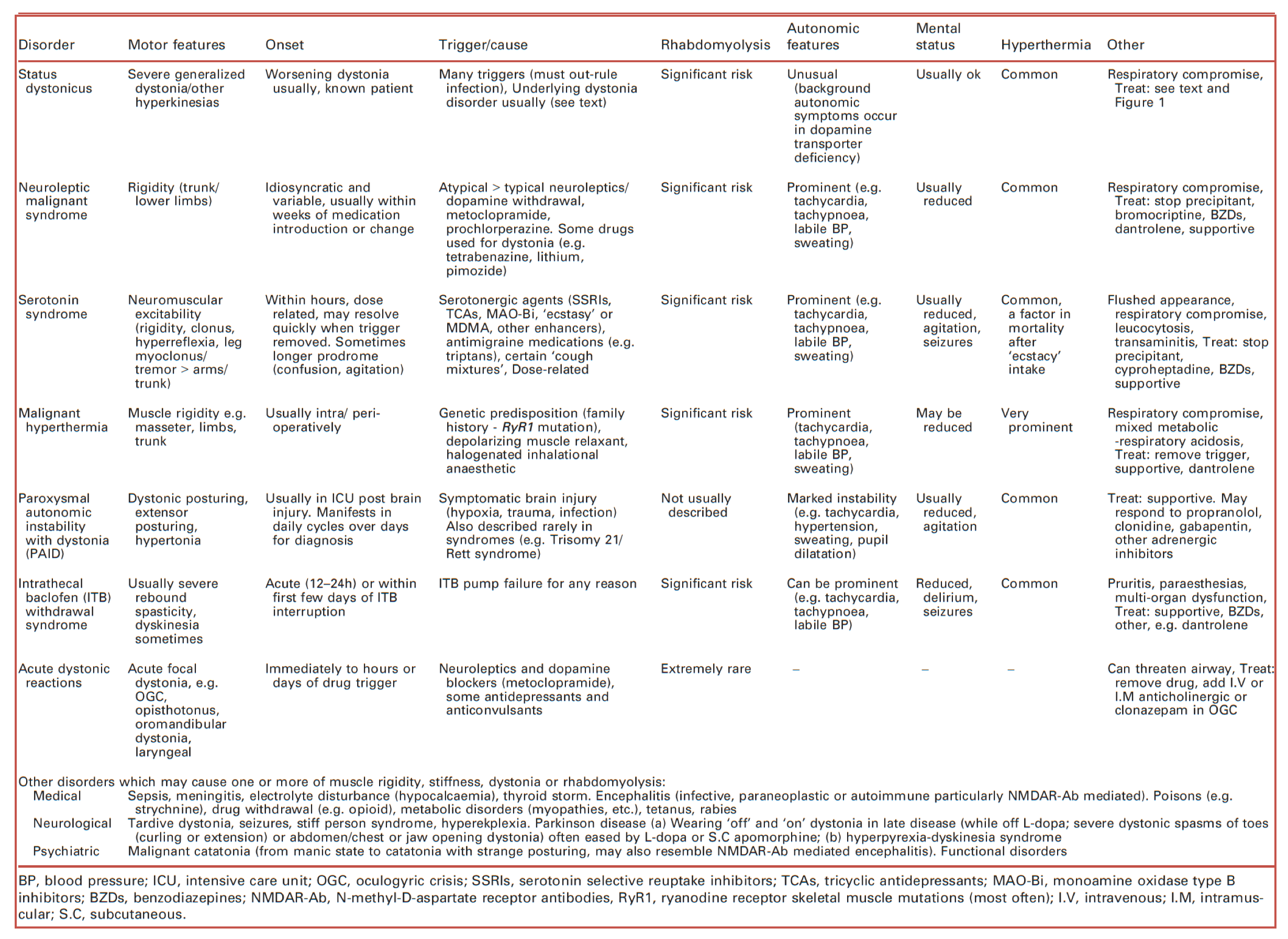
🔸The following table provides distinguishing features of some malignant movement disorders *.
▪️Evaluation
- Complete blood count, basic electrolytes, creatinine, BUN, calcium, magnesium, and phosphorus.
- Hepatic transaminases, lactate dehydrogenase, alkaline phosphatase.
- Creatinine kinase level.
- Urine myoglobin.
- TSH (if features of possible thyrotoxicosis).
- Sepsis workup (Blood culture, urinalysis, urine culture, lumbar puncture, CXR).
- Blood and urine toxicology screen.
- Brain imaging (CT, MRI) to exclude structural brain abnormalities.
- Electroencephalography (to rule out non-convulsive status epilepticus).
▪️Diagnostic criteria
- An international multispecialty consensus group published diagnostic criteria for NMS in 2011 *.
- These are based on positive clinical and laboratory findings as well as the exclusion of alternative causes, and each item is given a priority score for its relative importance in contributing to the diagnosis; however, no threshold score has been defined and validated for making a diagnosis of NMS.
-
- These criteria require independent validation before their use can be recommended in clinical practice. Other published criteria differ in requiring all or just some of the cardinal clinical and laboratory features.
- NMS diagnostic criteria: Expert panel consensus
- Exposure to dopamine antagonist, or dopamine agonist withdrawal, within the past 72 hours: 20 points
- Hyperthermia ( >38.0°C on at least 2 occasions, measured orally): 18 points
- Rigidity: 17 points
- Mental status alteration (reduced or fluctuating level of consciousness): 13 points
- Creatinine kinase elevation at least four times the upper limit of normal: 10 points
- Sympathetic nervous system lability, defined as at least two of the below: 10 points
- Blood pressure elevation (systolic or diastolic >25% above baseline)
- Blood pressure fluctuation (>25% systolic or >20% diastolic change in 24 hours)
- Diaphoresis
- Urinary incontinence
- Hypermetabolic state (defined as heart rate increase >25% above baseline and respiratory rate increase >50% above baseline): 5 points
- Negative evaluation for other toxic, metabolic, infectious, or neurologic causes: 7 points
- A validation study found that a cutoff of at least 74 points total (out of 100) could be used to determine the presence of NMS (with 70% sensitivity and 90% specificity, compared to expert adjudication) *.
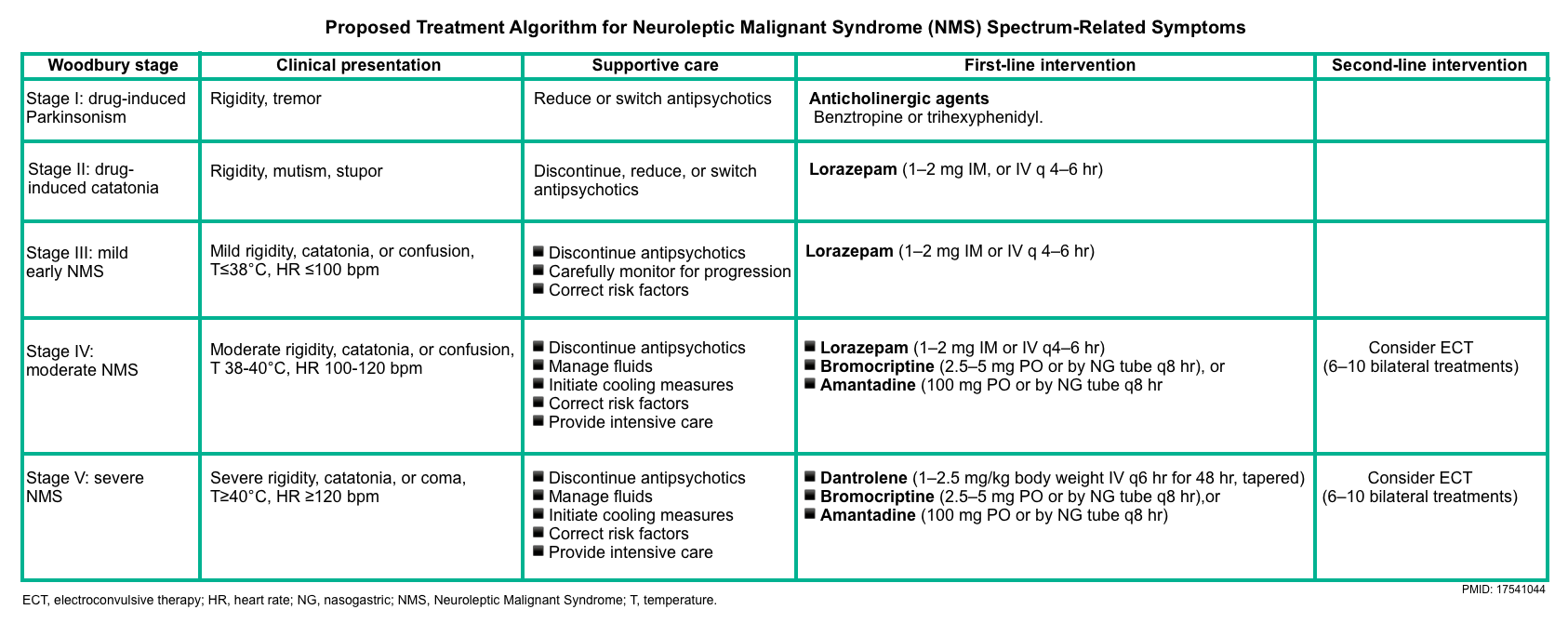
▪️Management
- Supportive care
- Discontinue dopamine antagonist therapy (or resume dopamine agonist in case of withdrawal of Parkinson’s medications immediately).
- Stop potential contributing agents (lithium, anticholinergic therapy, serotonergic agents).
- Treat hyperthermia with cooling blankets; ice water gastric lavage and axillary ice packs if required. Note that antipyretics are not helpful.
- Rhabdomyolysis is common. For the treatment see 📖.
- Monitor and correct electrolyte disturbances. Avoid nephrotoxins due to the high risk of renal failure.
- Maintain cardiorespiratory stability; cardiorespiratory monitoring; mechanical ventilation, antiarrhythmic drugs, and pacing may be required.
- Agitation
- Use benzodiazepines to control agitation, if necessary (however, they could cause or promote the development of delirium).
- For moderate to severe NMS, give benzodiazepines (e.g. lorazepam~2 mg IV q8hr) for agitation and muscle rigidity.
- Alternative options in patients with underlying psychosis:
- Central ⍺-2 agonists (clonidine, dexmedetomidine)
- Use benzodiazepines to control agitation, if necessary (however, they could cause or promote the development of delirium).
- Discontinue dopamine antagonist therapy (or resume dopamine agonist in case of withdrawal of Parkinson’s medications immediately).
- Specific treatments
- Bromocriptine is the first-line treatment.
- It is a dopamine agonist and is prescribed to restore lost dopaminergic tone. It is well tolerated in psychotic patients.
- NMS-specific therapy (e.g. bromocriptine and dantrolene) might reduce mortality in severe cases when compared to purely symptomatic therapy with benzodiazepines *.
- Doses of 2.5 mg (through nasogastric tube) q6-8h are titrated up to a maximum total dose of 40 mg/day. It is suggested that this be continued for 7 to 14 days after NMS is controlled and then tapered slowly.
- Alternative dopaminergic agent: amantadine can be used, if bromocriptine is contraindicated.
- For severe and refractory NMS, amantadine could be combined with bromocriptine *.
- Dantrolene is a second-line treatment.
- It is a direct-acting skeletal muscle relaxant that effectively treats malignant hyperthermia (MH).
- Doses of 1 to 2.5 mg/kg IV are typically used in adults and can be repeated to a maximum dose of 10 mg/kg/day.
- Efficacy includes reduction of heat production as well as rigidity, and effects are reported within minutes of administration.
- While some recommend discontinuing it after a few days, others suggest continuing for 10 to 14 days followed by a slow taper to minimize the risk of relapse
- ⚠️Contraindications & cautions:
- There is an associated risk of hepatotoxicity, and dantrolene should probably be avoided if liver function tests are very abnormal.
- The combination of dantrolene with calcium channel blockers is contraindicated.
- Electroconvulsive therapy (ECT)
- It is generally reserved for patients not responding to other treatments or in whom nonpharmacologic psychotropic treatment is needed.
- The rationale for the use of ECT in NMS includes its efficacy in treating malignant catatonia and reports of parkinsonism improving with ECT. A further impetus for ECT comes from the frequent need for psychotropic therapy in a setting in which antipsychotics cannot be used.
- Bromocriptine is the first-line treatment.
⛔️Anticholinergics are contraindicated in NMS.
Serotonin syndrome
▪️General
- Serotonin syndrome (SS) can be produced by any drug or, more commonly, by a combination of drugs that increase central serotonin neurotransmission.
- SS is a clinical diagnosis; no laboratory test can confirm the diagnosis.
- It may lead to death through the hyperthermia pathway. Hyperthermia may promote seizures, leading to a seizure-coma-death spiral.
▪️Cause of serotonin syndrome
- Generally, serotonin syndrome can happen for the following reasons:
- Overdose of a serotonergic medication (e.g. SSRI or illicit drugs). Serotonin syndrome occurs in ~15% of SSRI overdoses *.
- Inadvertent interaction between several serotonergic medications (see table below).
- Addition of directly serotonergic drugs.
- Addition of drugs that cause drug-drug interactions, increasing the levels of other serotonergic medications (i.e. the “new” medication doesn’t necessarily need to directly affect serotonin signaling). For examples:
- CYP3A4
- Inhibited by: ciprofloxacin or ritonavir.
- Metabolizes: methadone, venlafaxine, oxycodone.
- CYP2C19
- Inhibited by: fluconazole.
- Metabolizes: sertraline.
- CYP3A4
- Renal dysfunction may cause an accumulation of serotonergic medications (e.g. sertraline) *.
- Medications that can cause or promote serotonin syndrome *
- Psychiatric
- SSRIs (selective serotonin reuptake inhibitors): citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline
- SNRIs (serotonin-norepinephrine reuptake inhibitors): venlafaxine, duloxetine, milnacipran, and desvenlafaxine
- DNRIs (dopamine-norepinephrine uptake inhibitors): bupropion
- TCAs (tricyclic antidepressants): amitriptyline, amoxapine, clomipramine, desipramine, doxepin, imipramine, maprotiline, nortriptyline.
- Antidepressants/mood stabilizers: mirtazapine, trazodone, and lithium (increased serotonin release).
- MAOIs (monoamine oxidase inhibitors): safinamide, selegiline, rasagiline, phenelzine, tranylcypromine.
- Second-generation antipsychotics that are strong antagonists at 5-HT2A receptors and might indirectly activate 5-HT1A receptors and thereby promote serotonin syndrome: quetiapine, risperidone, olanzapine, clozapine, and aripiprazole.
- Buspirone.
- Antiemetics: ondansetron, metoclopramide.
- Antiepileptics: carbamazepine, valproate.
- Triptans: sumatriptan, and zolmitriptan.
- Ergot derivatives: ergotamine and methylergonovine.
- Opioids: levorphanol, meperidine, methadone, fentanyl, oxycodone, pentazocine, pethidine, tapentadol, tramadol, dextromethorphan.
- Substance use: cocaine, amphetamine, methamphetamine, MDMA, LSD.
- Miscellaneous:
- Linezolid (inhibits MAO)
- Methylene blue (inhibits MAO)
- Cyclobenzaprine (a skeletal muscle relaxant)
- Antihistamines: chlorpheniramine (inhibition of 5-HT uptake from synaptic cleft).
- Psychiatric

▪️Clinical presentation
- Chronocity
- It usually occurs shortly (within 2-24 hours) after exposure to medication or dose adjustment.
- It will rapidly (within 24 hours) resolve following the discontinuation of causative medications *.
- Clinical features
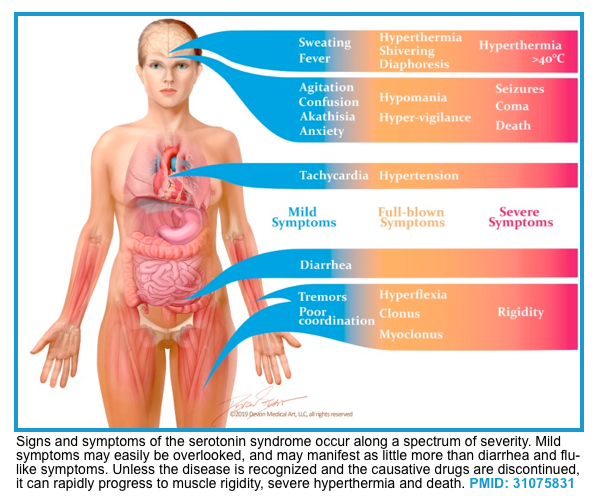
- The clinical presentation of serotonin syndrome represents a spectrum with mild serotonin toxicity at one end to a fulminant SS at the other (shown below).
- The triad of cognitive, autonomic, and neuromuscular effects is a classic feature of the serotonin syndrome.
- The most frequently noted clinical features include:
- Altered mental status, agitation, coma.
- Hyperreflexia, clonus, myoclonus, tremors, incoordination.
- Diaphoresis, shivering, and fever.
- GI: Nausea, vomiting, diarrhea.
- The most frequently noted clinical features include:
- Considerations
- Patients with ataxia should be examined carefully for lower extremity hypertonia.
- Unilateral muscle rigidity and focal neurologic findings are not expected.
- ⚡️Seizures are always generalized and usually short-lived.
- 🌡Hyperthermia is usually of moderate severity, but temperatures >41°C have been reported and are a marker of a poor prognosis.
- Hypertension is twice as common as hypotension and is associated with a more favorable prognosis.
- Patients with ataxia should be examined carefully for lower extremity hypertonia.
▪️Diagnosis
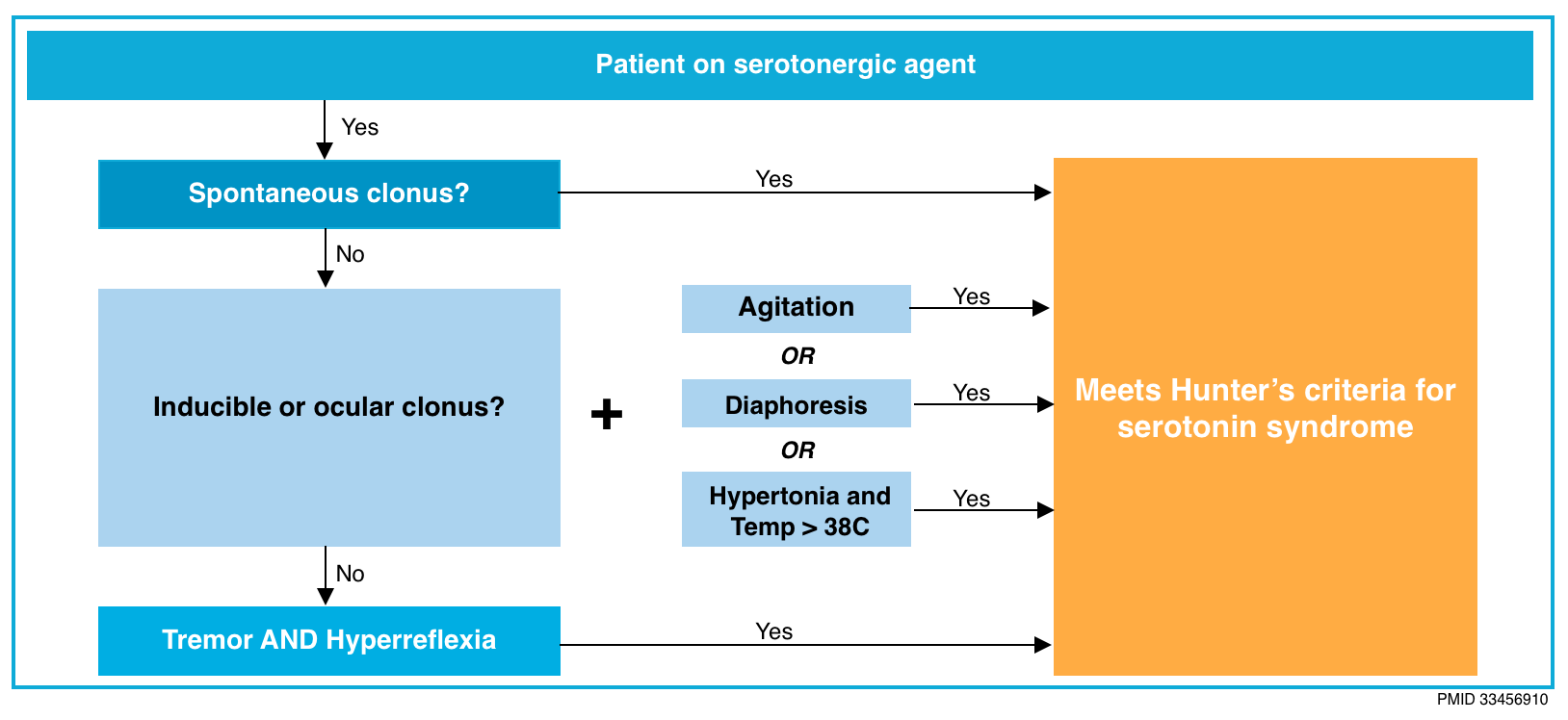
- The Hunter Criteria for SS are fulfilled if the patient has taken a serotonergic agent AND has one of the following:
- Spontaneous clonus
- Inducible clonus and agitation or diaphoresis
- Ocular clonus and agitation or diaphoresis
- Tremor and hyperreflexia
- Hypertonia
- Temperature >38°C and ocular clonus or inducible clonus
▪️Management
- Principles
- Discontinue serotonergic agents.
- SS is self-resolving promptly following the discontinuation of causative agents.
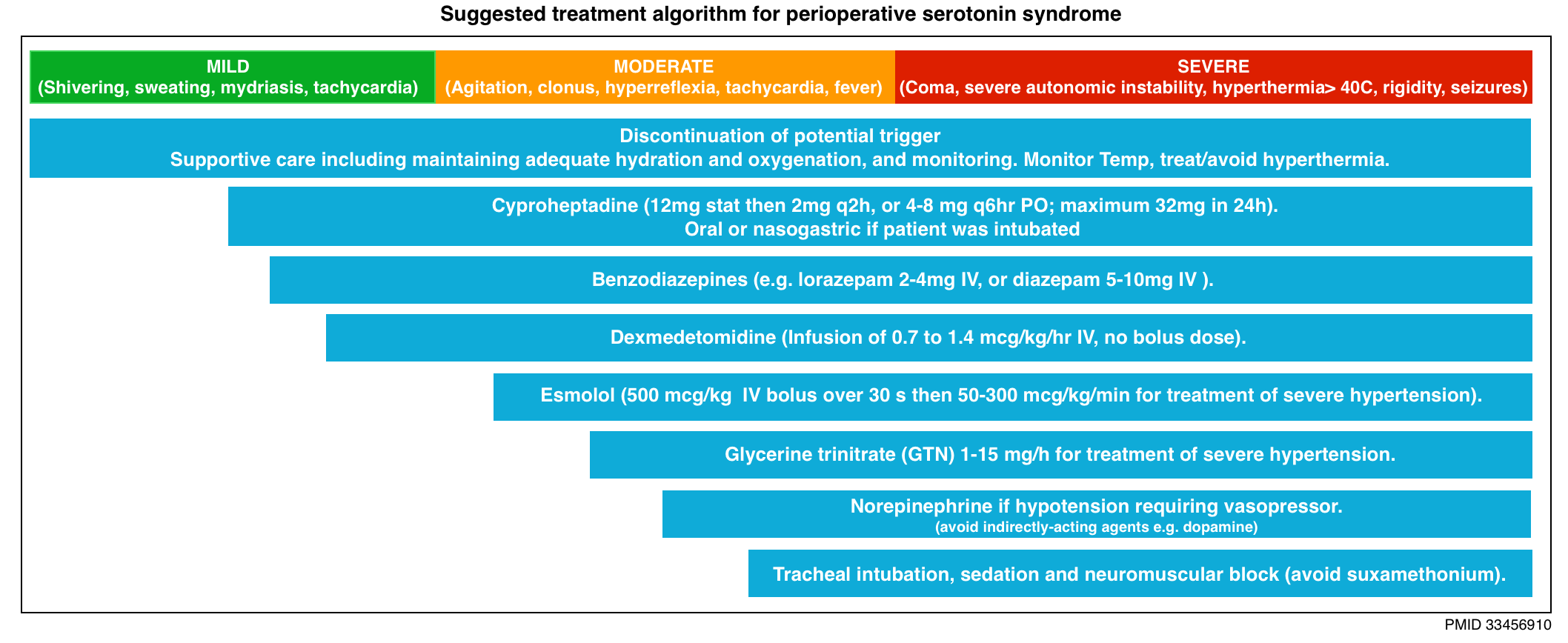
- The type and intensity of management depend on the clinical manifestations of SS (below algorithm).
- Patients manifesting more than mild symptoms should be closely monitored in a high-dependency care environment (Level 2 or 3).
- The mainstays of treatment for all presentations of SS include:
- Discontinuation of serotonergic agents.
- Supportive care
- Management of agitation, hyperthermia, and autonomic instability.
- Discontinue serotonergic agents.
- Supportive care
- Supportive care should aim to restore normal physiology in all cases.
- Provide oxygen (maintain SpO ≥94%); IV fluids; continuous cardiac monitoring.
- Anticipate complications; in severe SS vital signs can fluctuate widely and rapidly.
- Monitor temperature and treat/avoid hyperthermia.
- Treatment of agitation 😡
- Sedation should be used only if the patient is dangerously/uncomfortably agitated or hyperthermic.
- Cyproheptadine
- It is an H1 receptor antagonist that also has 5-HT1A and 5HT2A receptor antagonist activity (most closely related to serotonin syndrome).
- Disadvantages
- Only available orally.
- Absorption may take some hours.
- Side effects include sedation, hypotension, and anticholinergic effects (e.g. tachycardia, urinary retention).
- ⚠️Anticholinergic effects could exacerbate hyperthermia.
- When to give cyproheptadine in SS?
- For patients with very mild agitation who can take oral medication, cyproheptadine may be tried.
- Dosing: Loading dose of ~12 mg. Maintenance dose of 4-8 mg q6hr (16-24 mg total daily dose).
- Contraindications: Narrow-angle glaucoma, bladder obstruction.
- Benzodiazepine
- For patients with severe agitation who require immediate sedation or had a seizure.
- For an intubated patient, consider dexmedetomidine or propofol rather than benzodiazepines.
- Dexmedetomidine
- It stimulates alpha-2A receptors in the prefrontal cortex and locus coeruleus, which causes sedation and reduced sympathetic tone. It may work centrally to modulate serotonin levels.
- Advantages of dexmedetomidine in comparison to benzodiazepines:
- It doesn’t suppress respiration, so it is less likely to precipitate intubation.
- It can be titrated to effect, thereby avoiding over-sedation or under-sedation.
- It is less likely than benzodiazepines to exacerbate delirium.
- Disadvantages of dexmedetomidine in comparison to benzodiazepines
- It lacks anti-epileptic activity.
- Up-titration of dexmedetomidine takes some time, so it may not be an ideal agent for a patient with profoundly dangerous agitation.
- When to give dexmedetomidine in SS?
- It can be given to patients with more severe agitation who require more immediate sedation and have no prior seizure.
- Dosing 💉
- Do not give boluses of dexmedetomidine as it may cause bradycardia and hemodynamic collapse.
- Rather, the infusion may be started at a high rate (e.g. 1-1.4 mcg/kg/min) and down-titrated as the drug takes effect (within an hour).
- Infusion rate at 0.7 to 1.4 mcg/kg/hr.
- Management of Hyperthermia
- Principle
- Mild hyperthermia
- It can be controlled with physical cooling techniques (e.g. cooling blanket, fan), and benzodiazepine sedation to decrease muscle activity.
- Severe uncontrolled hyperthermia (temperature >41.1C)
- This can lead to severe complications, such as rhabdomyolysis, metabolic acidosis, and DIC.
- Aggressive treatment with immediate sedation, paralysis, endotracheal intubation; and neuromuscular block with non-depolarising agents is necessary to reduce muscle activity and control body temperature.
- 💡Consider intubation in the following conditions
- Status epilepticus.
- Extreme hyperthermia (Temp > 41C).
- Chest wall rigidity interfering with ventilation.
- Uncontrollable agitation.
- ⛔️Avoid
- ⚠️Succinylcholine as this may worsen rhabdomyolysis. Nondepolarizing agents are preferred (e.g. rocuronium).
- ⚠️Fentanyl, since it may promote serotonin syndrome.
- Management of autonomic instability
- Hypertension and tachycardia appear to be far more prevalent than hypotension.
- Hypertension
- Use short-acting, rapid titratable medications such as esmolol or nitroglycerine *.
- ⚠️Avoid hydralazine, as it has significant inhibitory effects on MAO and may exacerbate serotonin toxicity.
- Hypotension
- IV fluids, and vasopressors (e.g. norepinephrine, epinephrine, phenylephrine) may be required.
- ⚠️Avoid indirectly acting catecholamines, such as dopamine and ephedrine, especially when agents that inhibit the activity of MAO are involved. In the presence of MAO inhibition, the effects of indirect amines can be unpredictable.
- Hypertension
- Hypertension and tachycardia appear to be far more prevalent than hypotension.
Malignant hyperthermia (MH)
▪️General
- It results from exposure to halogenated inhalational anesthetics or depolarizing muscle relaxants e.g. succinylcholine in a genetically susceptible individual.
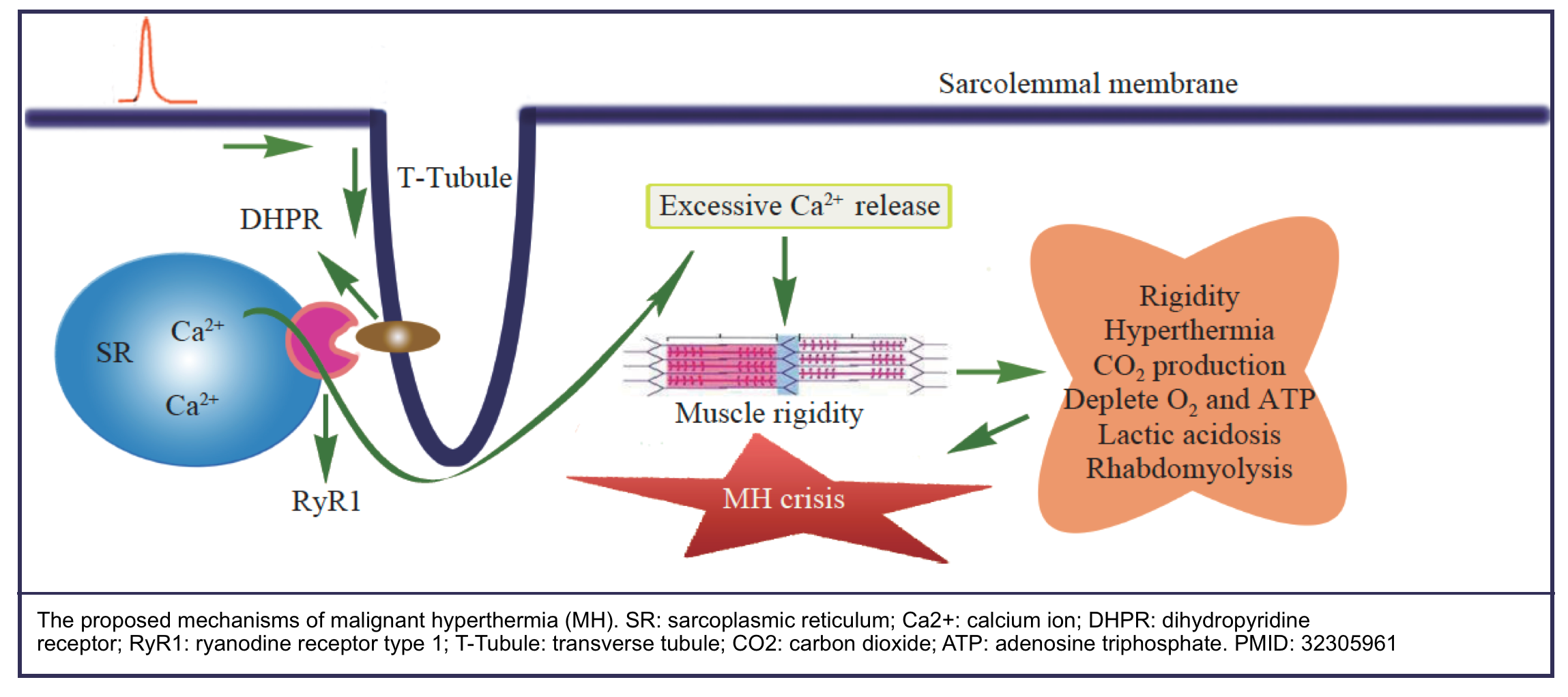
- The underlying mechanism of MH is excessive calcium release from the sarcoplasmic reticulum (SR) which leads to disturbance of intracellular calcium ion (Ca2+) homeostasis and uncontrolled skeletal muscle hypermetabolism *.
- This hypermetabolic state generates heat and leads to hypercarbia, hypoxemia, acidosis, arrhythmias, rhabdomyolysis, renal and circulatory failure, and
fatal outcome.
- This hypermetabolic state generates heat and leads to hypercarbia, hypoxemia, acidosis, arrhythmias, rhabdomyolysis, renal and circulatory failure, and
▪️Clinical presentation
- Chronocity: MH is classically fulminant and acute, although some insidious forms have been reported.
- Clinical features
- Hypercarbia, hyperthermia, autonomic instability, rigidity, and rhabdomyolysis.
- Increased carbon dioxide production (92%) is the hallmark of malignant hyperthermia since MH is a muscle-based hypermetabolic process *.
- Tachycardia (73%) is another early sign of MH. Tachycardia combined with hypercarbia is highly suspicious of a hypermetabolic state.
- Hyperthermia (52%) is a key indicator of MH, but it can be a late sign or may be absent *.
- Generalized muscle rigidity (~65%) is caused by muscle contraction.
- Rhabdomyolysis is a late sign of MH and can result in life-threatening hyperkalemia.
- Myoglobinuria from rhabdomyolysis may also lead to acute renal failure.
- Other findings
- Masseter spasms (~27%).
- Alteratered consciousness.
- Electrolyte imbalances.
- Cardiac arrhythmias (mostly due to hyperkalemia).
- Hypoxemia.
- Pulmonary edema, and congestive heart failure.
- Hypercarbia, hyperthermia, autonomic instability, rigidity, and rhabdomyolysis.
▪️Differential diagnosis
- Sepsis, and anaphylaxis.
- Inadequate anesthesia and/or analgesia *.
- Anesthesia machine dysfunction *.
- Other disorders to consider in all cases with a hypermetabolic state presentation (tachycardia, hyperthermia, hypercarbia, hemodynamic instability) include *:
- Thyrotoxicosis, pheochromocytoma, neuroleptic malignant syndrome, serotonin syndrome, and intoxication (e.g. cocaine, and ecstasy).
- Unlike in NMS, metabolic acidosis and hypercapnia are more prominent, and transaminitis is less likely to occur *.
- Thyrotoxicosis, pheochromocytoma, neuroleptic malignant syndrome, serotonin syndrome, and intoxication (e.g. cocaine, and ecstasy).
▪️Evaluation and monitoring during treatment
- Core temperature, ECG, and continuous vital monitoring.
- End-tidal CO2.
- ABG/VBG..
- Creatinine kinase, serum & urine myoglobin.
- Renal function tests, and electrolytes.
- INR, PTT, and fibrinogen.
- Sepsis workup (if suspected of infections).
- Check the glucose level hourly if insulin is administered (for hyperkalemia) *.
▪️Diagnosis
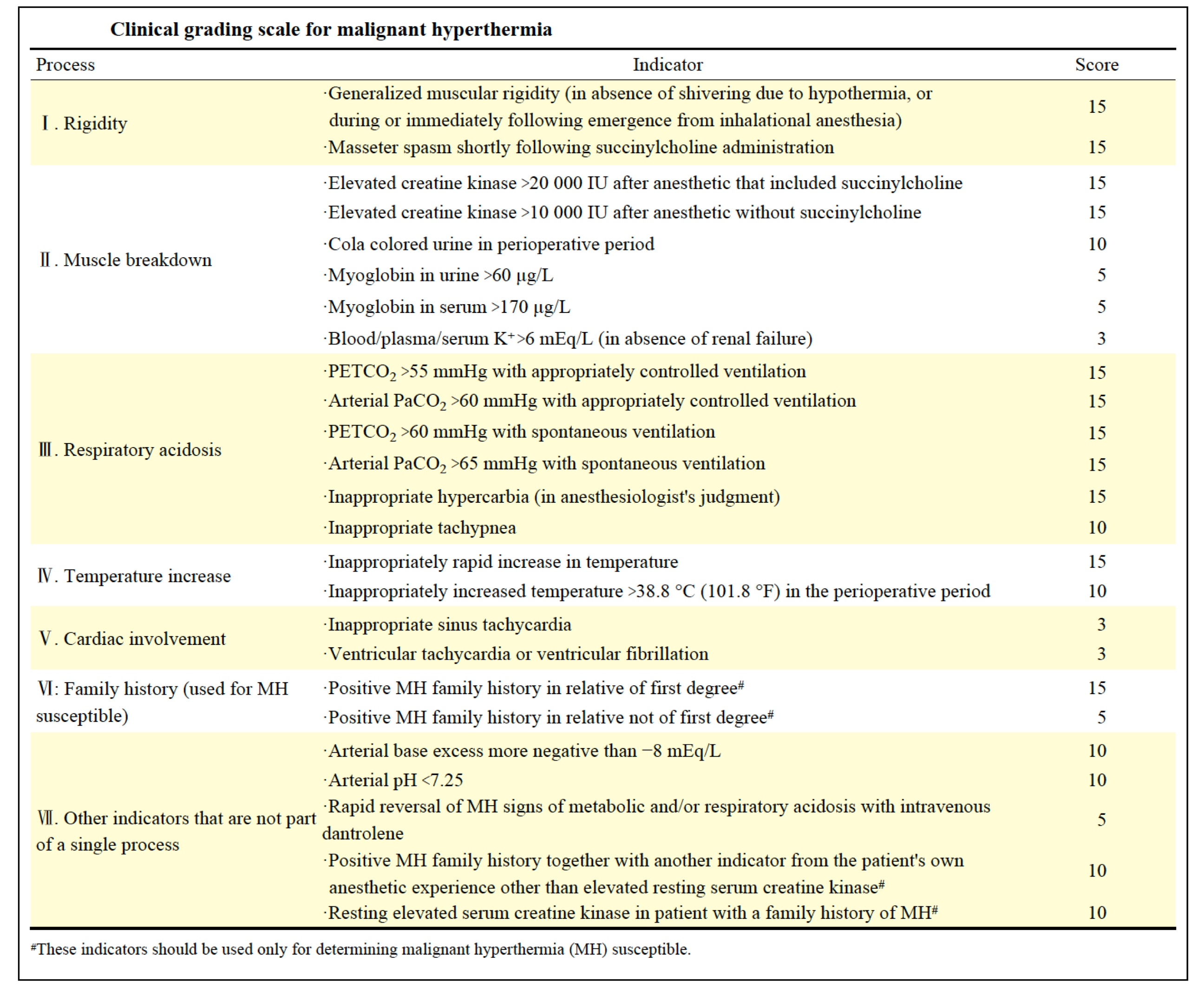
- The diagnosis of MH is based on clinical symptoms and laboratory testing.
- The following clinical grading scale can be used to qualitatively estimate the likelihood of an MH event and MHS.
- MH is likely to occur when the score goes more than 20, while MH may almost be clinically diagnosed when the score goes more than 50.
▪️Treatment
- Supportive care
- Discontinue inhaled anesthetics and succinylcholine.
- Hyperventilate (use a minute volume 2–3 times normal) with 100% oxygen at flows of 10 L/minute; to clear any residual volatile anesthetic.
- Increase the minute ventilation. Titrate the respiratory rate and tidal volume to achieve an end-tidal pCO2 of ~25 mmHg *.
- Hyperventilate with 100% oxygen, and perform endotracheal intubation if ETT is not in place *.
- Treat hyperkalemia in patients with arrhythmias or potassium >6 mEq/L. 📖
- Treat arrhythmias per ACLS.
- ⛔️Avoid calcium channel blockers (which may interact with dantrolene *).
- Most arrhythmias respond to the correction of hyperkalemia and acidosis.
- Cool the patient as necessary: Start cooling for core temperature >39°C, and discontinue cooling when temperature decreases to 38°C *.
- Surface cooling with wet, cold sheets, fans, and ice packs placed in the axillae and groin.
- Administer cool or cold intravenous isotonic crystalloid.
- Management of rhabdomyolysis (more on this 📖)
- Isotonic bicarbonate (D5W with three 50-mEq ampules of bicarbonate per liter) might be the fluid of choice for resuscitation as this may be beneficial for hyperkalemia, acidosis, and rhabdomyolysis.
- Specific treatment
- Dantrolene
- It is a muscle relaxant drug that acts as an antagonist at the ryanodine receptor to slow the release of calcium and to allow the cells to reincorporate it into the sarcoplasmic reticulum.
- It is well tolerated clinically, The most commonly described side effect is muscle weakness in patients who received high doses of dantrolene.
- Loading dose: 2.5 mg/kg given as IV rapid bolus.
- Watch for reversal of clinical signs (ETCO2 should begin to normalize, vital signs stabilize, and rigidity ends) *.
- Repeat dantrolene bolus (2.5 mg/kg IV) as necessary; cumulative doses ≥10 mg/kg IV may be required (up to 30 mg/kg).
- Maintenance dose: After the initial MH event is controlled, administer dantrolene 1 mg/kg IV q4-6h or 0.25 mg/kg/h for at least 24 hours.
- Dantrolene
Catatonia
▪️General
- Catatonia is a motor dysregulation syndrome involving difficulty initiating or terminating actions *.
- Although catatonia is most commonly seen in patients with major depression disorder, bipolar disorder, and schizophrenia; it can occur in patients without such a history, often in association with acute nonneurological illness.
- Therefore, catatonia should not be assumed to be due to a psychiatric diagnosis, and appropriate evaluation should be done to exclude an underlying medical or neurological cause.
- Catatonia is a spectrum disorder combining bizarre features (e.g. mutism, posturing, echolalia, see below) with variable degrees of severity.
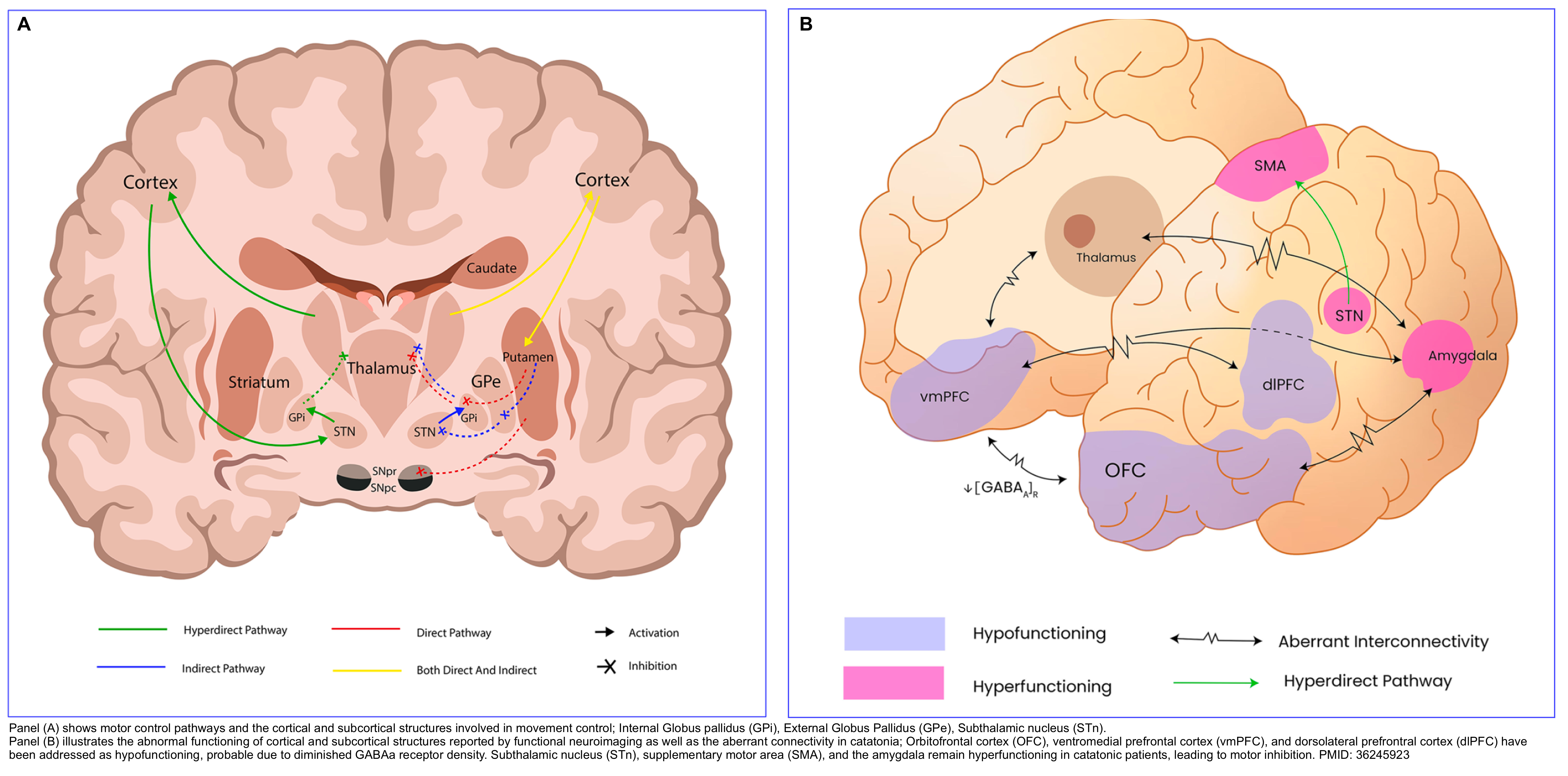
▪️Pathophysiology
- One helpful model for conceptualizing catatonia is to think of it as a basal ganglia disorder, with lesions in the basal ganglia thalamocortical tracts and the anterior cingulate/medial orbitofrontal circuit.
- The neurotransmitter imbalance may involve:
- ⬇️Dopamine activity at D2-receptors.
- ⬇️GABA signaling.
- ⬆️Signaling via glutaminergic NMDA receptors *.
▪️Clinical features (🎥)
- Catatonia is a spectrum disorder that includes some combination (≥3) of the following *,*
- Mutism: Verbally unresponsive, without evidence of aphasia.
- Stupor: Absence of movement or other reaction to stimulus while awake.
- Negativism: Contrary resistance to instructions or directions (does the opposite of what is requested)
- Posturing: Spontaneous and active maintenance of a posture against gravity
- Ambitendency: The patient seems to get stuck in indecisive movements.
- Cataplexy: Maintenance of fixed postures for prolonged periods with minimal movement regardless of stimuli, including pain.
- Waxy flexibility: Initial resistance followed by a slow release when the examiner moves the patient, like bending a warm wax candle.
- Gegenhalten: Resistance to movement with force equal and opposite that applied by the examiner
- Mitgehen: Movement in the direction of very slight pressure, despite an instruction to the contrary
- Echolalia: Mimicking the examiner’s speech
- Echopraxia: Mimicking the examiner’s movements. 🎥
- Automatic obedience: Exaggerated cooperation with a request or excessive continuation of a requested movement
- Mannerisms: Repetitive, odd, idiosyncratic movements or gestures unique to the individual
- Grimacing: Maintenance of odd facial expressions.
- Stereotypy: Repetitive movement or behavior
- Verbigeration: Continuous repetition of the same words or phrases
- Impulsivity: Suddenly behaves inappropriately for no apparent reason, unable to explain behavior.
▪️Types of catatonia
- Generally, catatonia can be described as either hypokinetic (stuporous) or hyperkinetic (excited).
- Both subtypes, if severe enough, can cause malignant catatonia which is characterized by autonomic instability.
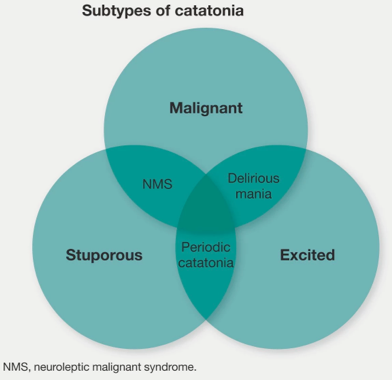
- Hypokinetic catatonia (causing stupor) 🎥
- This form is much more common than the hyperkinetic type *.
- Common features include immobility, staring, reduced interactivity with the environment, and mutism *.
- Rigidity and posturing may occur but are less common.
- Differential diagnosis of hypokinetic catatonia:
- Locked-in syndrome
- Nonconvulsive status epilepticus
- Psychogenic coma
- Abulia and akinetic mutism
- Hypoactive delirium
- Extrapyramidal/movement disorders (e.g. Parkinson’s disease, acute dystonia, tardive dyskinesia).
- Hyperkinetic catatonia (causing agitation) 🎥, 🎥
- Common features include mannerisms, stereotypies, grimacing, echolalia, echopraxia, verbigeration, and combativeness *.
- Differential diagnosis of hyperkinetic catatonia:
- Hyperactive/agitated delirium
- Nonconvulsive status epilepticus, complex partial seizure (more on seizure 📖).
- Acute mania.
- Akathisia.
- Malignant catatonia (causing autonomic instability)
- Either hypokinetic or hyperkinetic catatonia, if severe enough, may lead to autonomic instability defined as malignant catatonia.
- The key finding is catatonia plus autonomic instability including:
- Hyperthermia (this is required for the diagnosis of malignant catatonia).
- Diaphoresis, labile blood pressure, and/or tachycardia, tachypnea.
- Other common findings in malignant catatonia:
- Rigidity is a central feature (80%).
- Altered consciousness.
- Differential diagnosis of malignant catatonia (MC)
- Neuroleptic malignant syndrome (NMS) is a subtype of malignant catatonia that is induced by neuroleptics *.
- CNS disease
- Meningitis or encephalitis.
- Hypothalamic disease (e.g. CVA or hemorrhage).
- Other causes of hyperthermia
- Heat stroke.
- Toxicologic: Serotonin syndrome, malignant hyperthermia, sympathomimetic overdose, salicylate intoxication, anticholinergic intoxication.
- Sepsis.
- Endocrine: Adrenal crisis, thyroid storm, pheochromocytoma.
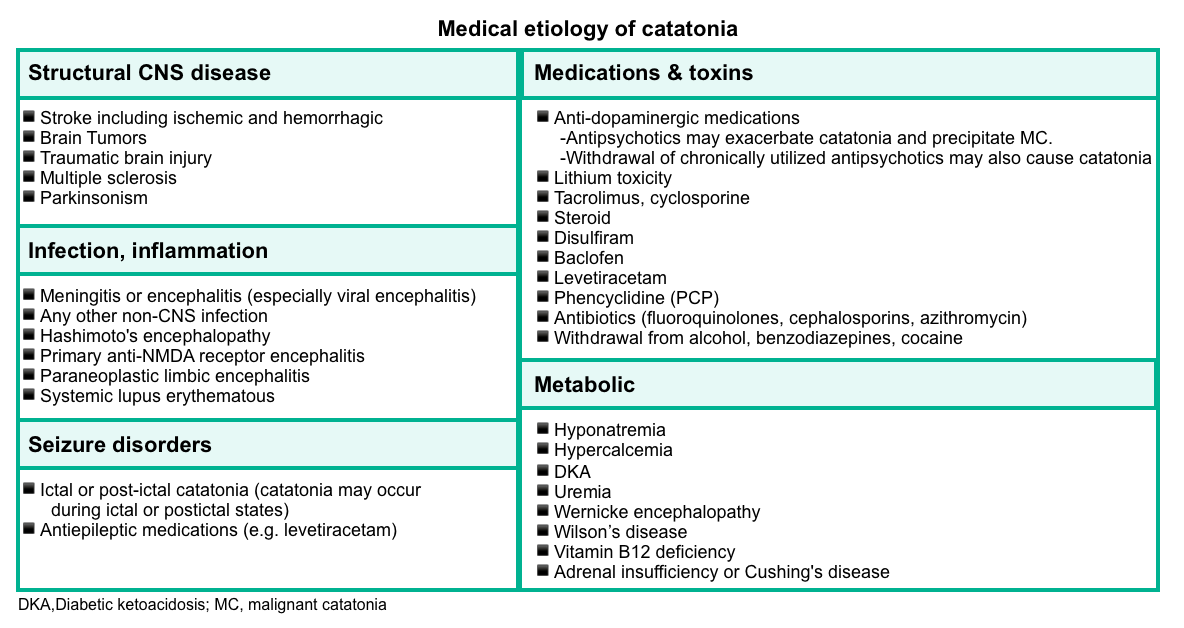
▪️Etiology
- Psychiatric
- Affective disorders are the most common psychiatric causes (especially bipolar disorder or depression with psychotic features).
- Psychotic disorders (including schizophrenia, schizoaffective disorder, and brief psychotic disorder due to acute stress).
- Autism spectrum disorder.
- Idiopathic catatonia
- Medical causes are summarized in the following table.
- Cell count with diff, renal and liver function tests, electrolytes.
- TSH level.
- Creatine kinase (to evaluate for rhabdomyolysis).
- Toxicologic screen (if appropriate).
- Lumbar puncture (if suspected of infection).
- Brain imaging.
- EEG
- EEG should be considered, especially in patients displaying bizarre behaviors (e.g. staring, grimacing, repeated movements). Some of these symptoms could occur in patients with complex partial seizures or nonconvulsive status epilepticus.
▪️Management
- Supportive care
- Treat the underlying cause.
- Removal of causative and/or perpetuating medication
- ⚠️Avoid antipsychotic medications. This may potentially worsen the condition leading to NMS/MC.
- 💡Withdrawal of chronic therapy with antipsychotics may precipitate catatonia. In this scenario, antipsychotic resumption may be helpful.
- 👉Restart dopamine agonists if these were recently discontinued (e.g. Parkinson’s medications).
- Management of hyperthermia and rhabdomyolysis.
- DVT prophylaxis especially among immobile patients.
- Nutritional support especially for patients who refused to eat for several days.
- Patients with malignant catatonia should be treated similarly as those with neuroleptic malignant syndrome including dantrolene for severe, intractable rigidity and hyperthermia, and short-acting antihypertensive medications for dysautonomia)
- Removal of causative and/or perpetuating medication
- Treat the underlying cause.
- Specific treatment
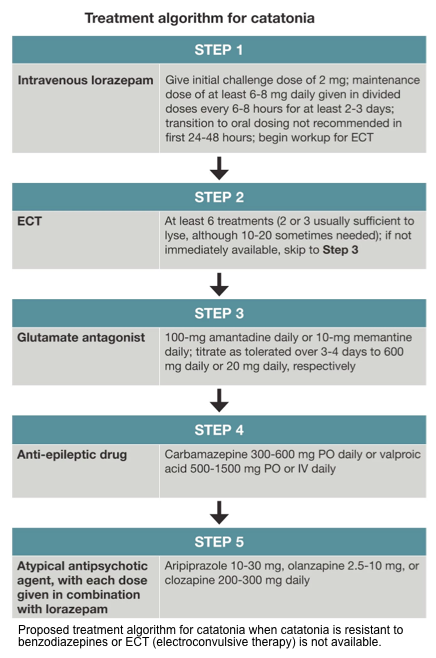
- Benzodiazepine is the front-line agent for catatonia (including both hypokinetic catatonia and hyperkinetic catatonia).
- The mainstay treatment for abatement of catatonic features is low-dose benzodiazepines (less than 8 mg/d of lorazepam).
- Electroconvulsive therapy (ECT)
- It should be considered for patients with refractory malignant catatonia or those with catatonia and inadequate response to medical treatment for ~2-5 days.
- The conventional course consists of daily treatment of up to 5 days followed by 3 times a week until improvement occurs.
- Alternative treatments (when benzodiazepines are ineffective and ECT is unavailable)
- Dopaminergic agents
- It should be considered especially if catatonia was precipitated by the use of antipsychotics (similar to the use of bromocriptine in NMS).
- ⚠️Caution is required in the context of underlying psychotic disorders, as dopaminergic agents can exacerbate the underlying psychosis.
- Carbidopa/levodopa may also be considered in refractory catatonia, at doses ranging from 25-100 mg/day *,*
- Memantine, amantadine
- Both agents inhibit NMDA receptor signaling, and amantadine could also augment dopamine signaling *.
- Amantadine: Start as 100 mg daily or BID. This may be increased over 3-4 days to a maximal dose of 200-300 mg BID *.
- ⚠️Caution in patients with a history of seizures.
- Memantine: Start as 5 mg BID. This may be increased over 3-4 days to 10 mg BID *,*
- Aripiprazole (10-30mg/d)
- It is an atypical antipsychotic with partial agonist activity at D2 receptors.
- It might be helpful to treat catatonia in the context of underlying psychosis *.
- Valproic acid (500-1500mg PO or IV daily): It is thought to be effective in catatonia by increasing GABA signaling.
- Dopaminergic agents
- Benzodiazepine is the front-line agent for catatonia (including both hypokinetic catatonia and hyperkinetic catatonia).
Parkinsonism
▪️Definition
- Parkinsonism refers to a constellation of motor disturbances that include (🎥):
- Hypokinesia (sometimes called bradykinesia or akinesia)
- This refers to slowness of voluntary movement and a reduction in automatic movements, such as swinging the arms while walking, a relatively immobile face (hypomimia or masklike facies) with infrequent blinking.
- The voice is soft (hypophonia) and poorly modulated.
- The handwriting is small (micrographia), tremulous, and hard to read.
- Rigidity.
- This is characteristic of Parkinsonism and is responsible for the flexed posture of many patients.
- The rigidity in Parkinsonism may manifest as cogwheel rigidity because of ratchet-like interruptions of passive movement that may be due, in part, to the presence of tremor.
- Tremor.
- Typically most conspicuous at rest; it increases at times of emotional stress and often improves during voluntary activity.
- It commonly begins with rhythmic, opposing circular movements of the thumb and index finger (“pill-rolling”); as rhythmic flexion–extension of the fingers, hand, or foot; or as rhythmic pronation–supination of the forearm.
- It frequently involves the lower jaw and chin as well.
- Although it may ultimately be present in all limbs, it is not uncommon for the tremor to be confined to one limb or both limbs on one side, for months or years before it becomes more generalized. In some patients, tremor never becomes prominent.
- Impaired posture and gait *.
- Hypokinesia (sometimes called bradykinesia or akinesia)

▪️Differential diagnosis of Parkinsonism
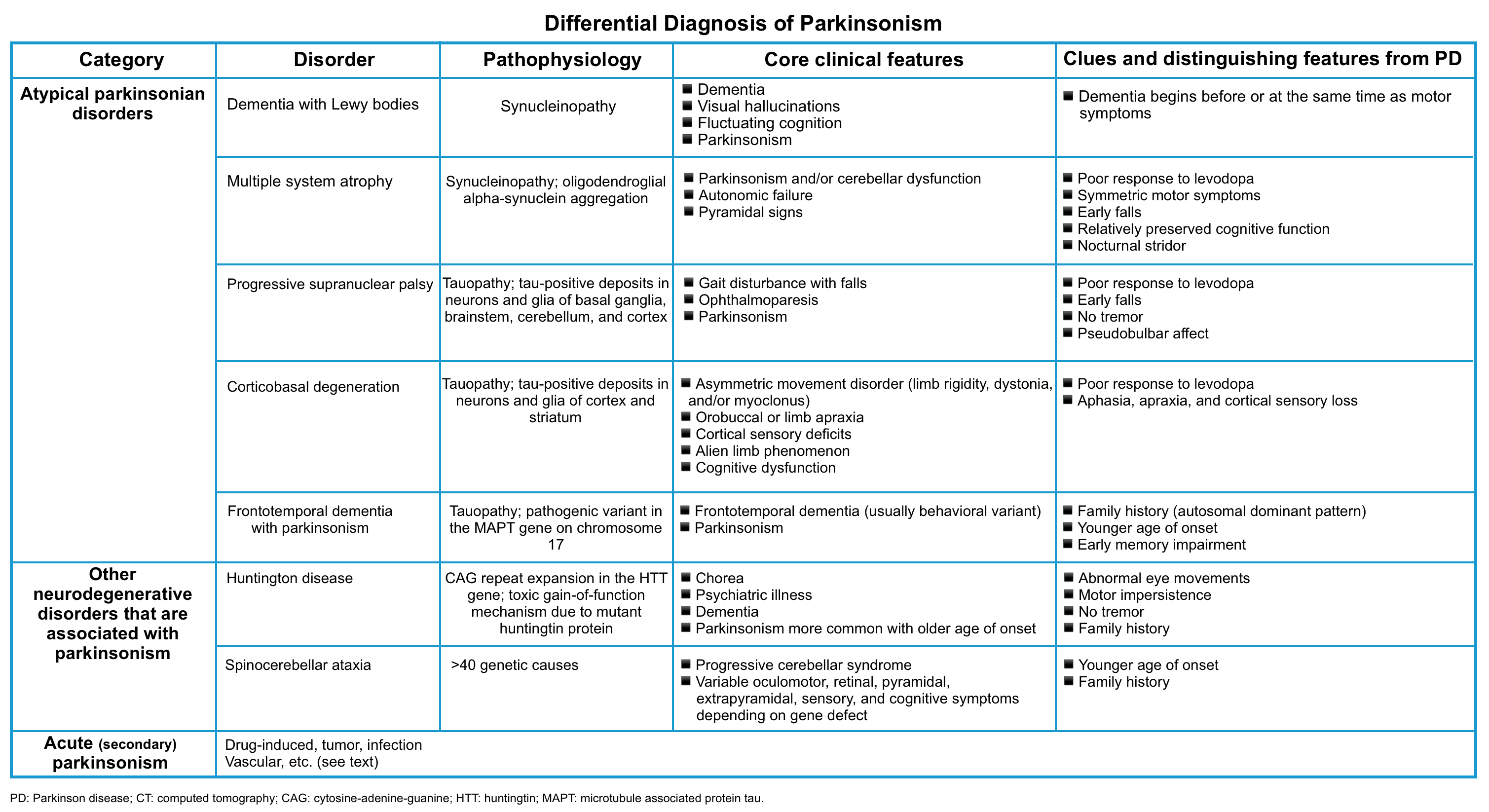
- Primary parkinsonism refers to parkinsonism resulting from a slowly progressive neurodegenerative condition such as Parkinson’s disease or atypical parkinsonian disorders (e.g. Lewy body disease, multisystem atrophy, progressive supranuclear palsy, and corticobasal degeneration) *.
- Acute parkinsonism evolves over hours to weeks from a variety of insults to the basal ganglia. This can be seen with several etiologies (below) *.
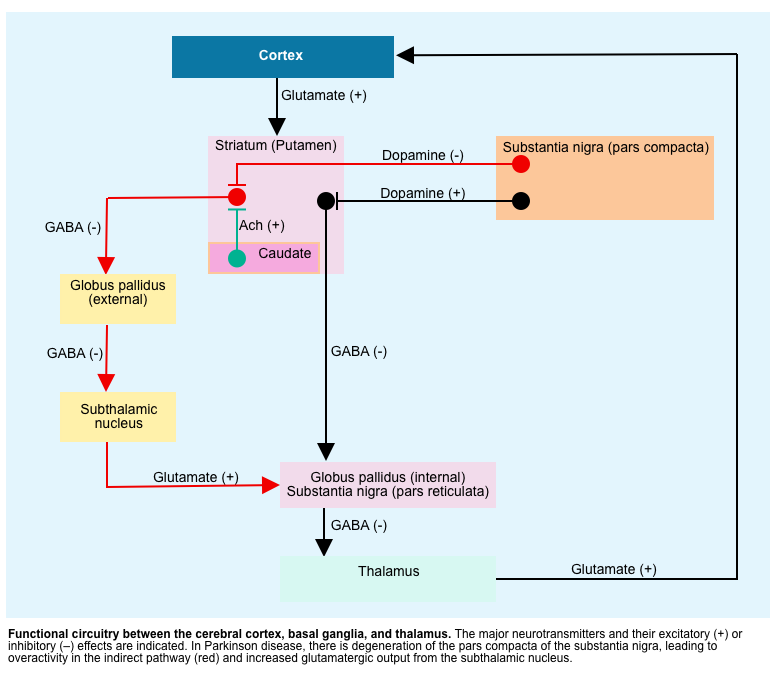
- The motor manifestations of Parkinsonism result from dysfunctional neurotransmitter pathways, mainly dopaminergic and cholinergic pathways within the basal ganglia *.
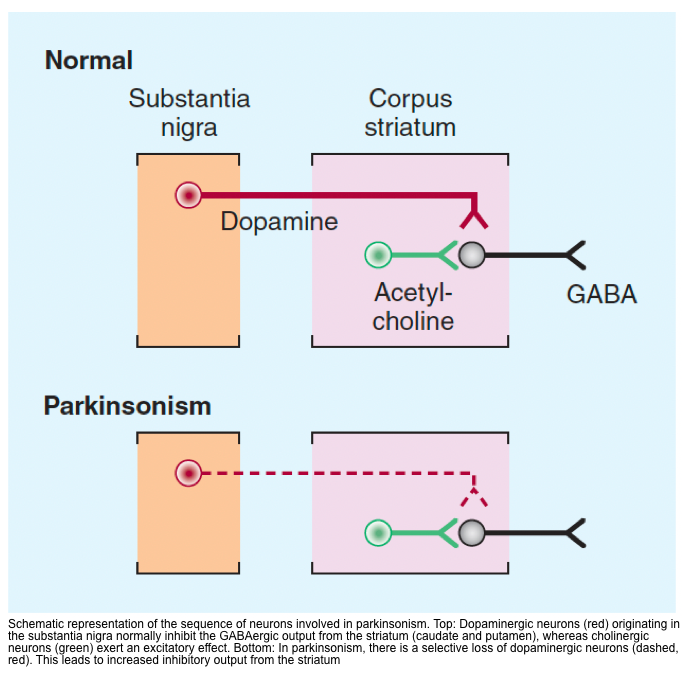
- In Parkinsonism, there is a selective loss of dopaminergic neurons in the substansia nigra leads to increased inhibitory output from the striatum {overactivity in the indirect pathway (red lines figure below)} *.
- In Parkinsonism, there is a selective loss of dopaminergic neurons in the substansia nigra leads to increased inhibitory output from the striatum {overactivity in the indirect pathway (red lines figure below)} *.
- ⬆️ Dopamine/Acetylcholine ratio can explain several movement disorders such as chorea/ballism
Acute Parkinsonism
▪️Etiology
- Medications
- Toxins
- Carbon monoxide, cyanide.
- Methanol, ethanol withdrawal; disulfiram.
- Manganese.
- Organophosphates.
- Structural brain lesions
- Stroke in basal ganglia, midbrain *
- Symptoms are often contralateral to the lesion and correspond to the vascular territory.
- Parkinsonism follows the initial phase of weakness.
- Cases of anterior cerebral artery strokes with contralateral hemiparkinsonism hypothesized as emerging from striatocortical circuit dysfunction have been reported *.
- Pontine myelinolysis.
- Obstructive hydrocephalus.
- Brainstem and posterior fossa tumors.
- Stroke in basal ganglia, midbrain *
- Infectious and inflammatory conditions
- Postencephalitic parkinsonism following various viral infections (EBV, HSV, CMV, VZV, HIV, influenza, poliovirus, measles, etc.).
- Systemic lupus erythematosus, antiphospholipid syndrome.
- Sarcoidosis.
- Brainstem encephalitis.
- Autoimmune or paraneoplastic inflammation brainstem.
- Metabolic
- Hepatic encephalopathy.
- Anoxic brain injury (may be delayed).
- Acute decompensation of Wilson’s disease.
- Central pontine myelinolysis.
▪️Clinical features
- Chronocity
- Signs usually develop within ~3 months after starting the offending drug and disappear over weeks or months after discontinuance.
- Clinical presentation (see Parkinsonism feature above).
- Hypokinesia tends to be symmetric and the most noticeable neurologic feature.
- Tremor is relatively uncommon.
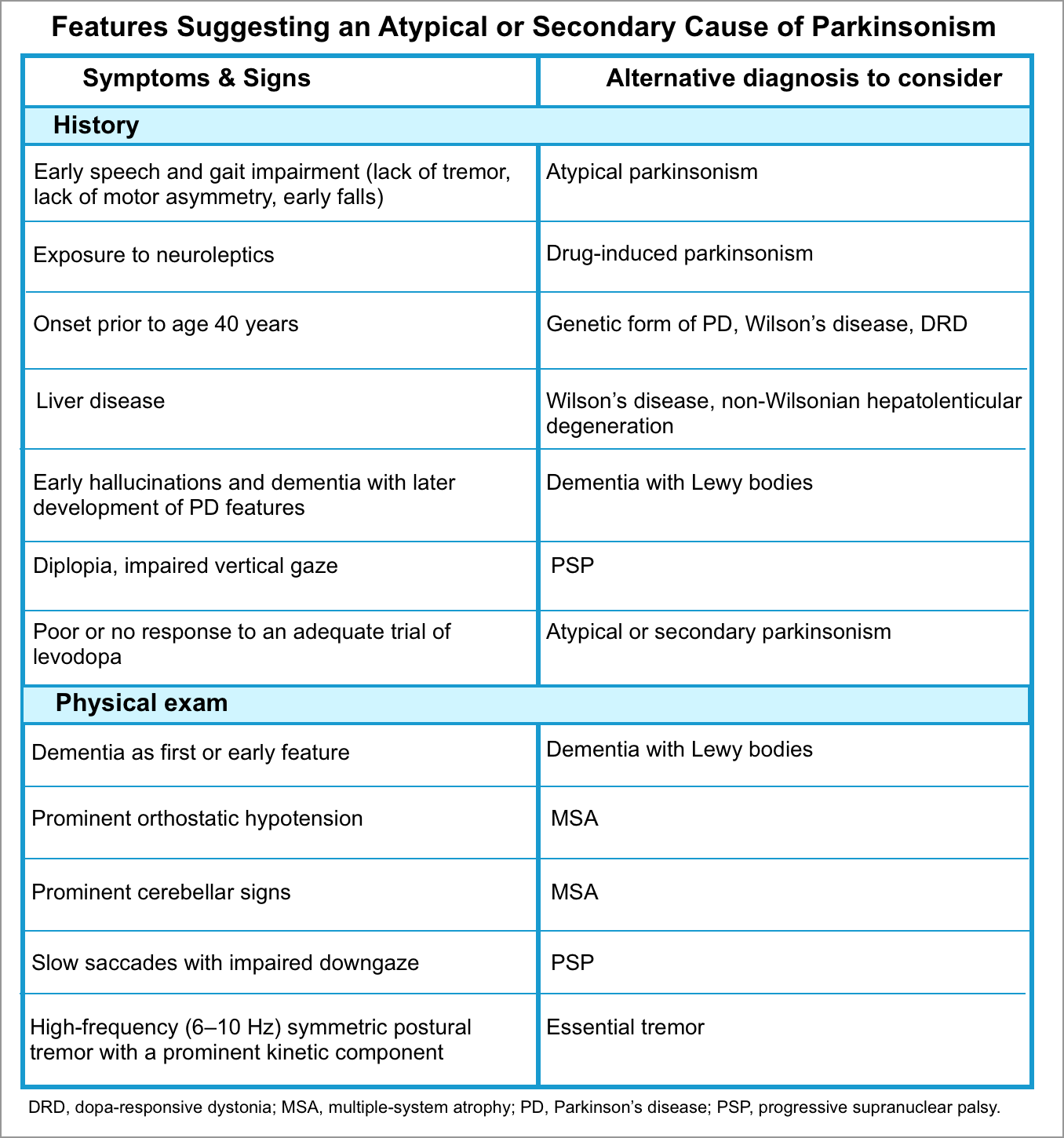
▪️Differential diagnosis
- Psychogenic (functional) parkinsonism.
- Tremors may show a jerky quality as opposed to rhythmicity, may disappear with movement of the limb, and often can be distracted and “entrained” *.
- Underlying Parkinson’s disease
- Patients with primary parkinsonism can develop “acute parkinsonism” as a drug reaction to dopamine-blocking agents such as antipsychotics (haloperidol), and antiemetics (prochlorperazine, metoclopramide).
- Acute extrapyramidal symptoms due to medications.
- Catatonia
▪️Pharmacology of antiparkinson drugs and other drugs used for selected movement disorders
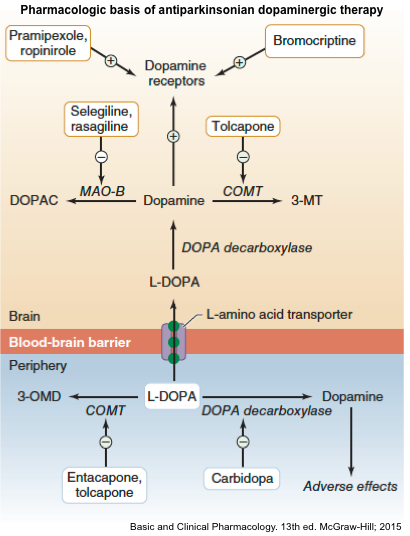
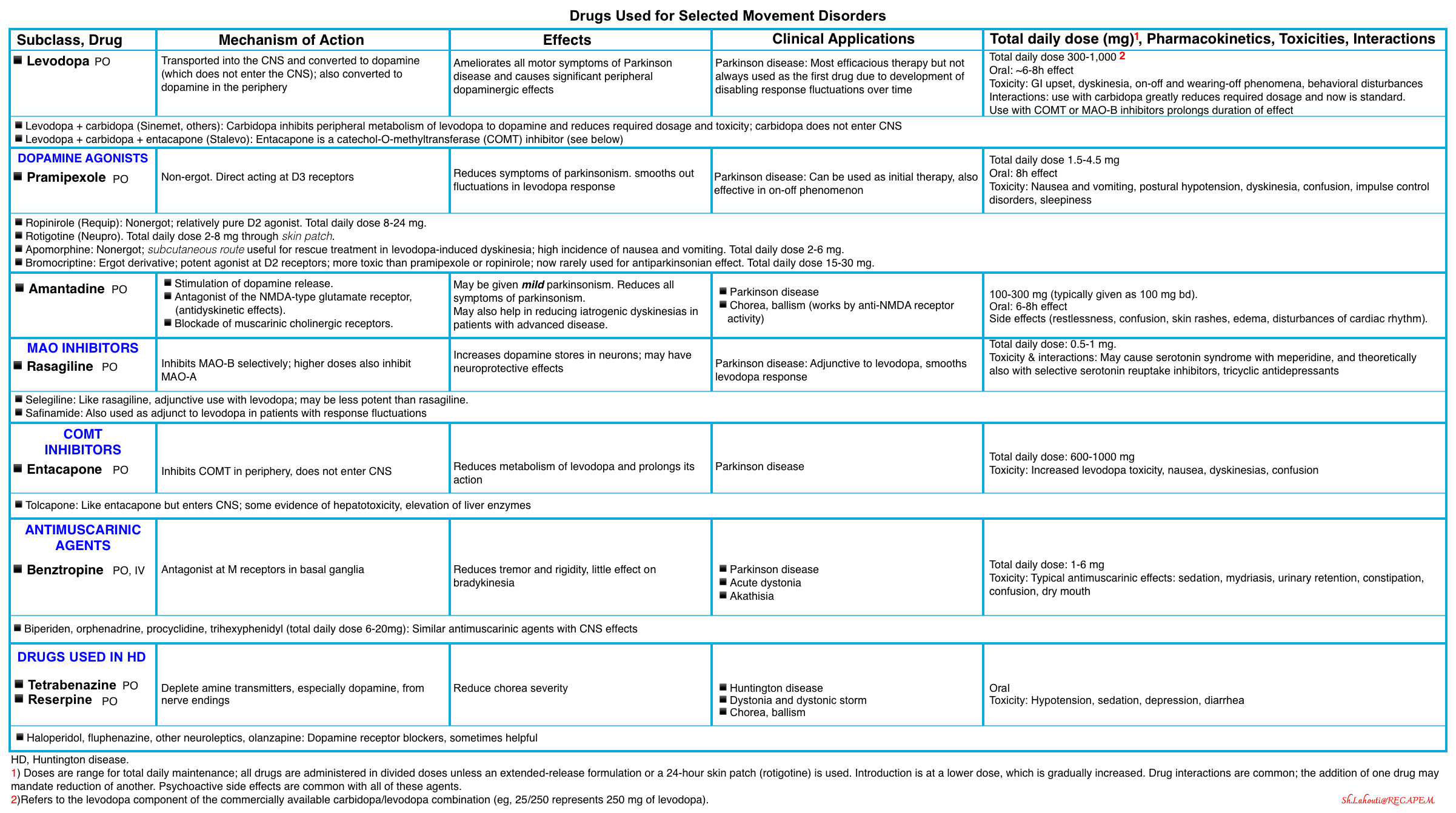
▪️Management
- Comprehensive workup about the suspected etiology, with appropriate interventions tailored to avoid recurrence.
- Symptomatic management
- A trial of levodopa (to target akinesia and rigidity) is better tolerated than postsynaptic therapies (dopamine agonists).
- Levodopa is recommended in all cases due to minimal associated side effects.
- If levodopa fails, amantadine 200-400 mg/day may be trialed.
- Once the primary pathology is managed, and symptoms improve in acute parkinsonism, dopaminergic therapy can be gradually weaned off.
- A trial of levodopa (to target akinesia and rigidity) is better tolerated than postsynaptic therapies (dopamine agonists).
Critical issues and emergencies in Parkinson’s disease
▪️Critical issues in the management of Parkinson’s disease
- The general principle of management of patients with Parkinson’s disease admitted for another reason in the hospital *
- Continue Parkinson’s medications, by and large, to avoid parkinsonism-hyperpyrexia syndrome.
- Avoid medications that inhibit dopamine transmission *
- ⛔️Antidopaminergics: Antipsychotics, antiemetics (e.g. metoclopramide, prochlorperazine, promethazine)
- Neuroleptics that can cause tardive dyskinesia must be used carefully, if at all, in patients with Parkinson’s disease.
- Ondansetron has less dopamine-blocking effects and is preferred over metoclopramide and prochlorperazine for nausea.
- ⛔️Medications that interact with levodopa
- Medications that decrease the effectiveness of levodopa: Pyridoxine, phenytoin, propoxyphene, papaverine, amitriptyline, isoniazid, iron.
- Decreases levodopa metabolism: Linezolid
- ⛔️Antidopaminergics: Antipsychotics, antiemetics (e.g. metoclopramide, prochlorperazine, promethazine)
- ⚠️Consider numerous drug-drug interactions of Monoamine oxidase-B inhibitors (MAO-B inhibitors; selegiline and rasagiline).
- In particular, they interact with a variety of medications to increase the risk of serotonin syndrome.
- MAO-B inhibitors should generally be continued, with caution regarding drug-drug interactions.
- Keep in mind that dopaminergic therapy toxicities can include(table above)
- Cardiac dysrhythmias.
- Orthostatic hypotension.
- Dyskinesias.
- Dystonias.
- Psychiatric and sleep disturbances, including nightmares, auditory and visual hallucinations, paranoia, and psychosis.
▪️Emergencies in Parkinson’s disease
- Although complications from Parkinson’s disease may develop over the disease course, sometimes unexpectedly, and require prompt or even urgent medical intervention. These emergent/urgent complications include *:
- Selected motor emergencies
- Severe levodopa-related motor complications
- Recurrent offs
- Dyskinesias (involuntary movements, e.g chorea, dystonia)
- Parkinson’s-Hyperpyrexia Syndrome
- Dyskinesia-hyperpyrexia syndrome
- Serotonin syndrome
- Severe levodopa-related motor complications
- Selected non-motor emergencies
- Psychosis in Parkinson’s disease
- Dysautonomic complications
- Symptomatic orthostatic hypotension and gastrointestinal tract complications, such as dysphagia and intestinal pseudobstruction.
- Selected motor emergencies
Severe levodopa-related motor complications
▪️Background
- Symptoms of Parkinsonism naturally fluctuate, and it is routine for patients to have periods where their symptoms are not optimally controlled.
- Usually, adjusting medication dosing or schedule corrects the problem, however, there are several caveats *:
- Parkinsonism worsens with medical illness, and it is not uncommon for urinary tract infections to present as a deterioration of parkinsonism, particularly in men who may not complain of frank dysuria.
- Marked deterioration in parkinsonism has been reported in several patients with subdural hematomas. Prompt recognition of this syndrome and surgical evacuation of hemorrhage is life-saving.
- Usually, adjusting medication dosing or schedule corrects the problem, however, there are several caveats *:
▪️Severe Levodopa-related motor complications
- Recurrent offs
- Motor fluctuations consisting of recurrent episodes of parkinsonism (off periods) of variable intensity that occur at different times to levodopa intake
(wearing off, delayed-on, no-on, unpredictable off) *. - Clinical features
- Severe tremors and unmanageable and painful freezing of gait, profuse sweating and tachycardia, abdominal discomfort, and also psychiatric manifestations, particularly depression, and anxiety *.
- Management
- Provide a more constant delivery of dopaminergic drugs to the brain, e.g. giving multiple doses of levodopa, or adjunctive treatments aiming to prolong the levodopa effect such as dopamine agonists, COMT inhibitors, and MAO-B inhibitors.
- Changing the levodopa dose throughout the day and adjusting the adjuvant medications can minimize the fluctuations and make them more predictable.
- However, about 10% of patients have severe medically refractory fluctuations; such patients require referral to specialized centers for consideration of device-aided therapies.
- Motor fluctuations consisting of recurrent episodes of parkinsonism (off periods) of variable intensity that occur at different times to levodopa intake
- Dyskinesias
- It can develop at different times in relation to the antiparkinsonian effect of levodopa (peak dose and diphasic dyskinesia, or off-period painful dystonia).
- Management
Psychosis in Parkinson’s disease
▪️Revised criteria for PD psychosis
- The diagnosis of Parkinson’s disease psychosis requires at least one of the following:
- Illusions.
- False sense of presence.
- Hallucinations.
- Delusions *.
- These features should occur following PD onset and should be present for at least 1 month, either as recurrent or continuous symptoms, excluding other causes (dementia or other psychiatric disorders such as schizophrenia or bipolar disorder) and noting associated features, such as the presence or absence of insight, dementia, and PD treatments.
▪️Epidemiology and risk factors
- Psychosis occurs in 20 to 40 % of drug-treated patients with PD, and visual hallucinations are the most common psychotic symptom *.
- Risk factors for psychosis in PD include:
- Antiparkinson drugs use:
- Psychosis may be provoked or exacerbated by Parkinson’s medications. Ordered from most problematic to least problematic, these include *: Anticholinergics > Amantadine > MAO-B inhibitors > Dopamine agonists > COMT inhibitors > Levodopa.
- High doses of antiparkinson drugs are used.
- Presence of dementia.
- Advancing age.
- Impaired vision, depression, presence of sleep disorders, high comorbid disease burden, and longer disease duration.
- Antiparkinson drugs use:
▪️Etiology of Psychosis in Parkinson’s Disease
- P—Parkinson’s disease medications
- SY—Systemic illness
- C—Centrally acting medication
- H—Hepatic, renal, or other metabolic dysfunction
- O—Overdose of medications or intoxication
- S—Sensory deprivation (hearing, visual impairment)
- I—Infection (urinary tract infection, pneumonia)
- S—Structural lesions (stroke, subdural hematoma, intracranial hemorrhage, trauma)
▪️Evaluation
- History/exams
- Review all medications with attention to recent changes in drugs or doses, or drug-drug interactions (e.g., serotonin syndrome).
- Adherence to medications?
- History of previous similar presentations, particularly urinary tract infections?
- Primary psychiatric conditions such as depression, schizophrenia, schizoaffective disorder, or bipolar disorder?
- Concomitant signs and symptoms?
- The presence of hallucinations with either delusion or confusion, agitation, or myoclonus suggests a toxic-metabolic encephalopathy *.
- These patients are medically ill, infected, or acutely overmedicated.
- Psychosis accompanied by dry skin, urinary retention, and mydriasis suggest anticholinergic toxicity *.
- The presence of hallucinations with either delusion or confusion, agitation, or myoclonus suggests a toxic-metabolic encephalopathy *.
- Temporal course
- For those PD patients with new-onset psychosis, hallucinations early in the course of parkinsonism (e.g. within the first 12 months or even before dopaminergic drugs are introduced) may indicate Dementia with Lewy bodies (DLB).
- This “1-year rule” has been used to separate DLB from PDD. This distinction has treatment implications as DLB patients may have marked neuroleptic sensitivity and, in rare cases, develop a neuroleptic malignant-like syndrome.
- Psychosis in DLB patients also may respond to cholinesterase inhibitor * .
- For those PD patients with new-onset psychosis, hallucinations early in the course of parkinsonism (e.g. within the first 12 months or even before dopaminergic drugs are introduced) may indicate Dementia with Lewy bodies (DLB).
- Investigations (Proposed tests, depending on scenario) *
- Labs: complete blood count, comprehensive metabolic profile, thyroid function, toxicology screen, urinalysis, urine culture, cerebrospinal fluid analysis.
- Imaging: head computed tomography or magnetic resonance imaging, chest X-ray.
- Other: electroencephalogram.
▪️Managment
- An overall view of the management of psychosis in PD is represented below.
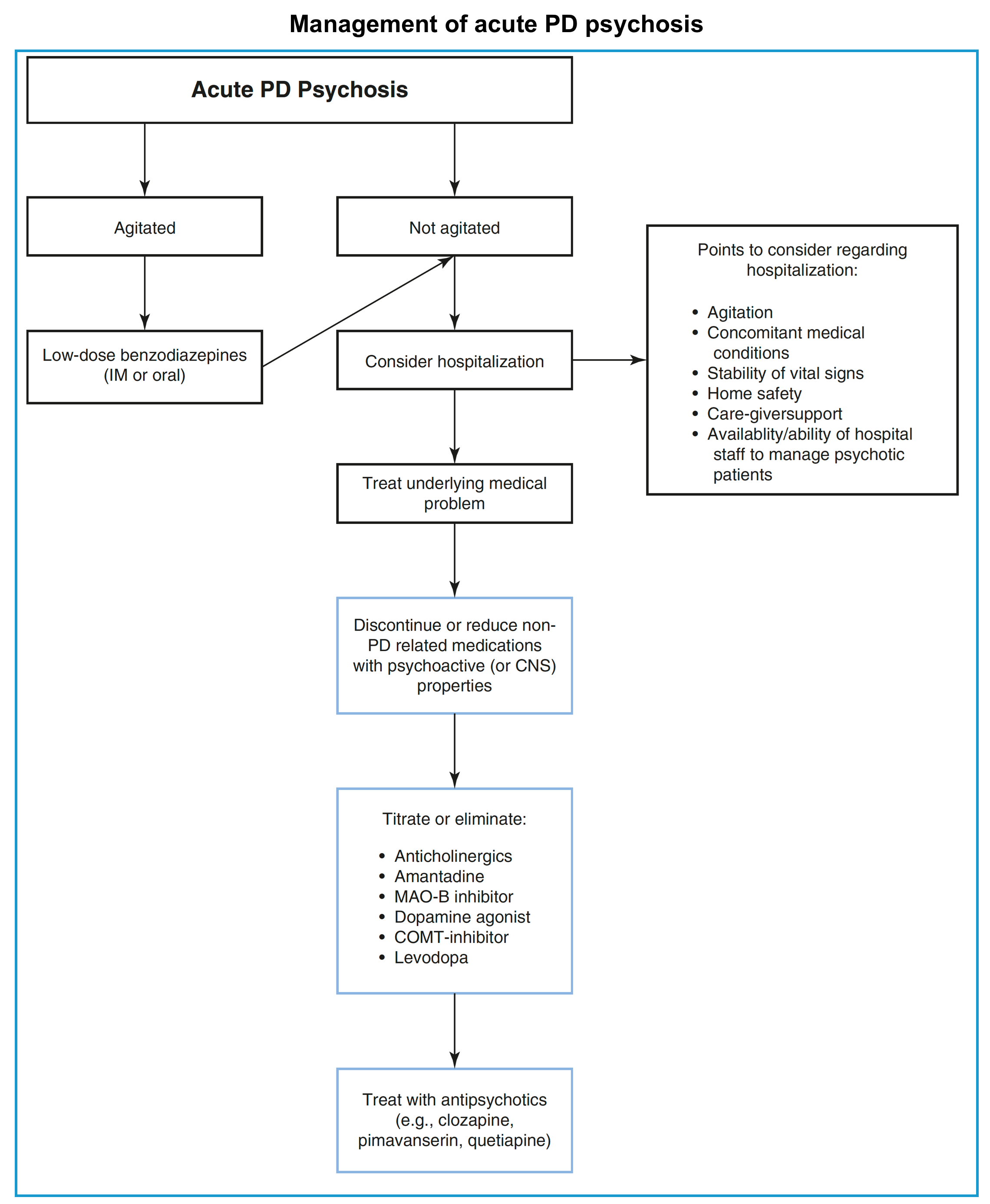
- Calm down the patient if severely agitated.
- In this situation, low-dose benzodiazepines (intramuscular or oral) may be reasonable.
- ⚠️ High-dose or long-term benzodiazepines should be avoided in this population.
- ⛔️Avoid typical antipsychotics with dopamine-blocking properties as they can trigger a significant deterioration of motor symptoms, and a neuroleptic malignant syndrome.
- In this situation, low-dose benzodiazepines (intramuscular or oral) may be reasonable.
- Identify and correct the possible underlying cause (dehydration, infection, and metabolic disorders are common).
- Discontinue or reduce non-Parkinson’s disease medications that may have psychoactive properties, including:
- Anticholinergics for bladder hyperactivity.
- Tricyclics for depression.
- Benzodiazepines for anxiety or sleep, hypnotics for sleep.
- Opioids for pain.
- Reducing Parkinson’s disease medications should be considered (especially, if there is no timely improvement following the above measures):
- Adjustment should be based on the potential of each medication to worsen psychosis.
- In general, reductions should first be made to adjunctive agents (anticholinergics, MAO-B inhibitors, amantadine) and then dopamine agonists and COMT inhibitors *.
- Reductions in levodopa should be made only if reductions of other antiparkinsonian agents have failed to be effective.
- ⚠️Care must be taken not to avoid removal of all dopaminergic medications, especially suddenly, because this can cause significant worsening of parkinsonism symptoms and lead to Parkinson-hyperpyrexia syndrome.
- There are no guidelines on how quickly one should reduce the dose of these medications, but changes have to be implemented cautiously and gradually, as motor symptoms can deteriorate significantly.
- It is always important to reduce polypharmacy first and then reduce levodopa gradually, based on the individual patient’s needs.
- Generally, do not reduce by more than 20% to 30% of levodopa in one interval.
- Specific therapy for psychosis
- When all other causes of Parkinson’s disease psychosis (PDP) have been addressed and the cause is therefore thought to be related to disease progression. A point, however, may be reached at which the PD medications cannot be reduced without compromising motor function, at which time giving antipsychotic medications is appropriate:
- Atypical antipsychotics (clozapine, pimavanserin, quetiapine).
- Pimavanserin
- It is approved for the management of psychosis in Parkinson’s disease. By acting predominantly at the 5-HT2A receptors, pimavanserin may have less risk of exacerbating Parkinson’s disease than other antipsychotics *.
- Quetiapine can be used alternatively, starting at 12.5 mg daily with gradual escalation to 50 mg daily at night or BID.
- Pimavanserin
- Cholinesterase inhibitors (donepezil, rivastigmine).
- They might improve neuropsychiatric features and behavioral problems associated with PDD and DLB.
- Drawback: These medications take longer to work and thus are not useful in acute psychosis, particularly in the emergency setting.
- Antidepressants (e.g., selective serotonin reuptake inhibitors, mirtazapine) can also be helpful in patients with PDP who are experiencing depression and/or insomnia symptoms.
- Atypical antipsychotics (clozapine, pimavanserin, quetiapine).
- When all other causes of Parkinson’s disease psychosis (PDP) have been addressed and the cause is therefore thought to be related to disease progression. A point, however, may be reached at which the PD medications cannot be reduced without compromising motor function, at which time giving antipsychotic medications is appropriate:
Parkinsonism-Hyperpyrexia Syndrome
- Parkinsonism-hyperpyrexia syndrome (PHS) is a form of neuroleptic malignant syndrome (NMS).
- It occurs in patients with PD who abruptly withdraw or reduce dopaminergic medications*.
▪️Causes
- Discontinuing dopaminergic medications, dose reduction, or non-adherence.
- Changing from immediate-release to extended-release formulations.
- Reduced absorption of levodopa due to coadministration with high-protein tube feeds.
- Discontinuing anticholinergics or COMT inhibitors (e.g. entacapone).
- Dysfunction of a deep brain stimulator (if the patient has a device it should be immediately interrogated).
- Infection or dehydration.
▪️Clinical presentation
- Course
- Symptoms may begin 18 hours to a week after changing therapy.
- Clinical features
- Severe rigidity and tremors develop first, with subsequent development of dysautonomia, agitation, delirium, and altered mental status (stupor, coma) in succession.
- Compared to other examples of NMS, extrapyramidal/parkinsonian features tend to be more pronounced and more routinely present (e.g. tremors, rigidity) *.
- Tachycardia is an early sign of dysautonomia.
- Fluctuating blood pressure, tachypnea, and sweating are also seen.
- Hyperthermia results from a combination of central thermoregulatory dysfunction and hyperactivity of muscles.
- Elevated CK and white cell counts (but are not as prominent as in NMS).
- Myoclonus and seizures are not uncommon.
- Severe rigidity and tremors develop first, with subsequent development of dysautonomia, agitation, delirium, and altered mental status (stupor, coma) in succession.
▪️Differential diagnosis
- Neuroleptic malignant syndrome
- A history of Parkinsonism and rapid change or discontinuation of dopaminergic medication is a clue for the diagnosis of PHS.
- Dyskinesia-hyperpyrexia syndrome
⭐️When a patient with Parkinson’s disease experiences acute hyperpyrexia, the PHS, DHS, and SS need to be considered *.
▪️Management
- PHS is a neurological emergency that requires treatment in an intensive care setting.
- Supportive care (see above)
- Specific treatment
- The reinstitution of dopaminergic therapy
- Keep in mind that gastrointestinal absorption of medications is often altered due to intestinal dysmotility in Parkinsonian patients.
- Levodopa
- Route: Since parenteral forms of antiparkinsonian medications are not widely available, nasogastric administration is recommended.
- Dosage: It should be started at the premorbid dose first and can be titrated.
- Additional treatments
- Dopamine agonists e.g. ropinirole, rotigotine, or apomorphine
- Bromocriptine 5 to 10 mg divided into three doses per day, titrated to effect, is recommended.
- The reinstitution of dopaminergic therapy
Baclofen withdrawal
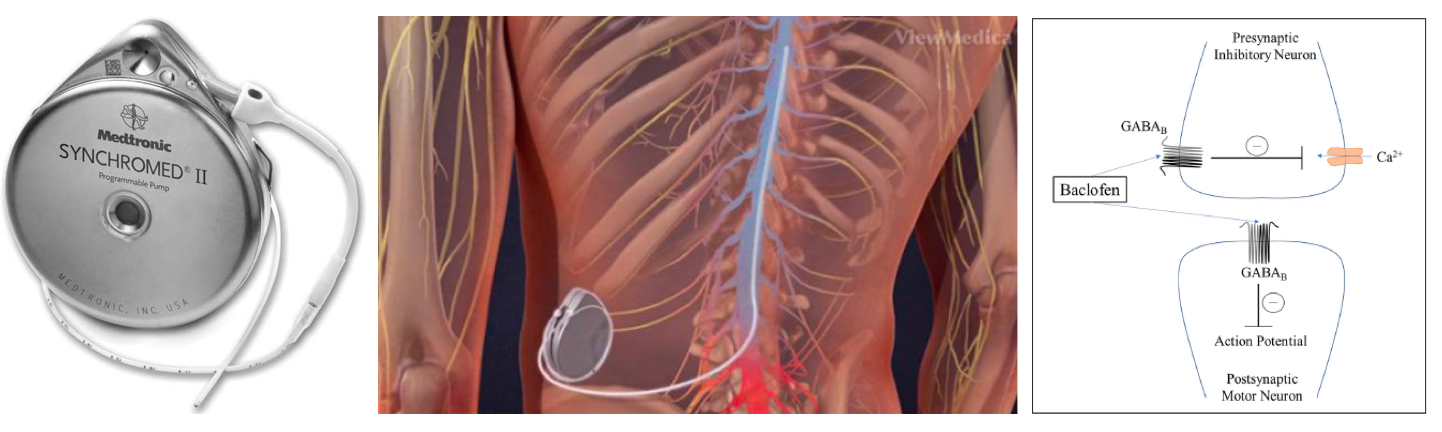
▪️Mechanism of action
- Baclofen is an agonist for gamma-aminobutyric acid (GABA)B receptors on pre- and postsynaptic neurons in the CNS and peripheral nervous system.
- GABA-B receptors have been found at the following sites:
- Brain, especially the cerebral cortex, thalamic nuclei, cerebellum, and amygdala.
- Ventral and dorsal horns of the spinal cord.
- Outside the blood-brain barrier, within the sympathetic nervous system and some visceral tissues (including the heart).
▪️Baclofen indications
- For spasticity of cerebral (e.g. cerebral palsy or traumatic brain injury) or spinal origin including management of spasticity associated with spinal cord injury and multiple sclerosis.
▪️Route of administration
- Oral
- Bioavailability: 75%–80%
- It distributes to both the brain (where it causes sedation) and also the spinal cord (where it causes muscle relaxation)
- Clinical effects of baclofen occur slowly (beginning over 3-4 days, with peak effect after 5-10 days), possibly due to slow penetration of the CNS.
- Baclofen’s water solubility does not allow it to readily cross the blood–brain-barrier (BBB) when administered orally. Accordingly, oral baclofen may require higher doses to achieve a therapeutic effect and have an increased potential for unwanted side effects, such as somnolence and sedation.
- Baclofen is excreted mostly by the kidneys, with a half-life of ~3 hours. Renal insufficiency may cause baclofen accumulation, with resultant toxicity.
- Intrathecal baclofen (ITB)
- Compared with oral administration ITB can more effectively treat spasticity directly at the level of the spinal cord with less associated systemic side effects.
- ITB administration allows for greater concentrations in the spinal cord (achieving greater muscle relaxation) without higher levels in the brain (which would cause sedation). Intrathecal baclofen has a half-life of ~5 hours
▪️Dosing
- Oral
- For treatment of spasticity, dosing starts at ~5 mg PO TID.
- The dose may be gradually escalated as tolerated (e.g. increasing by 5 mg/dose every 3 days), with a maximum dose of 80 mg/day.
- Acute ingestion of >200-400 mg baclofen is associated with a higher risk of coma, seizure, or intubation. However, patients on chronic baclofen therapy develop tolerance.
- Intrathecal baclofen
- The dose range extends from roughly 50 – 1,000 mcg/day.
- For spasticity of spinal cord origin, typical doses are 300 – 800 mcg/day.
- For spasticity of cerebral origin, typical doses are 90 – 700 mcg/day.
▪️Clinical manifestation and principles of management *
| System | Baclofen withdrawal | Baclofen toxicity |
| General | Pruritus Hyperthermia Multisystem organ failure, death | Hypothermia, death |
| Neurological | Altered mental status, delirium Hyperreflexia, myoclonus, hypertonia, rigidity. Tremors Rebound spasticity Paresthesias Headache Seizures | Hyporeflexia Tremor Impaired memory Confusion, lethargy, somnolence Seizures Encephalopathy Coma |
| Psychiatric | Hallucinations, anxiety, paranoia, delusions | Hallucinations, agitation, mania, catatonia |
| Musculoskeletal | Hypertonia, rhabdomyolysis | Hypotonia |
| Cardiovascular | Acute reversible cardiomyopathy, cardiac arrest Autonomic dysfunction: tachycardia, hypotension, hypertension | Conduction abnormalities, prolonged QTc Autonomic dysfunction: bradycardia, hypotension, Hypertension and tachycardia with high-dose toxicity |
| Respiratory | Respiratory failure | Respiratory failure |
| Gastrointestinal | Nausea, vomiting, diarrhea | Nausea, vomiting |
▪️Oral vs. ITB withdrawal
- Oral baclofen withdrawal is most often associated with the development of mild symptoms.
- This may be more notable for agitated delirium (given the distribution of oral baclofen to both the brain and spinal cord).
- ITB withdrawal is more likely to present with a severe, life-threatening withdrawal syndrome.
- This may rapidly progress to a multiorgan failure state resembling neuroleptic malignant syndrome (with tachycardia, rigidity, hypotension, fever, seizures, and rhabdomyolysis).
- ITB withdrawal may cause more pronounced spasticity (since there is predominantly a deficiency of baclofen within the spinal cord).
▪️Differential diagnosis
- Sepsis
- Meningitis (which could relate to an infected pump).
- Neuroleptic malignant syndrome
- Serotonin syndrome
- Malignant hyperthermia
- Autonomic dysreflexia
- Sympathomimetic intoxication
- Alcohol withdrawal *
Evaluation
- Investigate possible alternative diagnoses e.g. sepsis *.
- Plain radiography for patients with baclofen pumps may sometimes reveal catheter kinking, migration, or disconnection (with a series of films including several views). Baclofen pumps should also be interrogated to determine if they are functioning properly. More sophisticated imaging may be needed (e.g. with contrast injection into the pump’s catheter).
- Causes of ITB withdrawal:
- Pump malfunction.
- Dosing error.
- Depletion of the pump’s battery or baclofen reservoir.
- Intrathecal catheter migration or fracture.
- Intentional pump removal due to infection.
- Causes of ITB withdrawal:
- Investigation of the duration and timing of baclofen administration may be helpful.
- Baclofen withdrawal should occur within ~1-4 days of stopping oral baclofen (or <48 hours after stopping intrathecal baclofen, although the precise timing of pump malfunction may be unknown).
- If the patient was started on baclofen within <1 month, this would make baclofen withdrawal unusual.
▪️Management of baclofen withdrawal
- General supportive care
- Hemodynamic support
- Management of rhabdomyolysis
- Management of hyperthermia
- Fluid resuscitation due to insensible losses.
- Prevention and treatment of CNS complications such as seizures and delirium.
- Diminishing acute exacerbations in muscle spasticity *.
| Hyperkinetic Movement Disorders |
Hyperkinetic movement disorders refer to abnormal involuntary movements that typically do not include those related to convulsions, neuromuscular dysfunction (fasciculations), or reflex clonus.
These excess involuntary movements can be regular and rhythmic, as in tremor; more sustained and patterned, as in dystonia; brief and random, as in chorea.
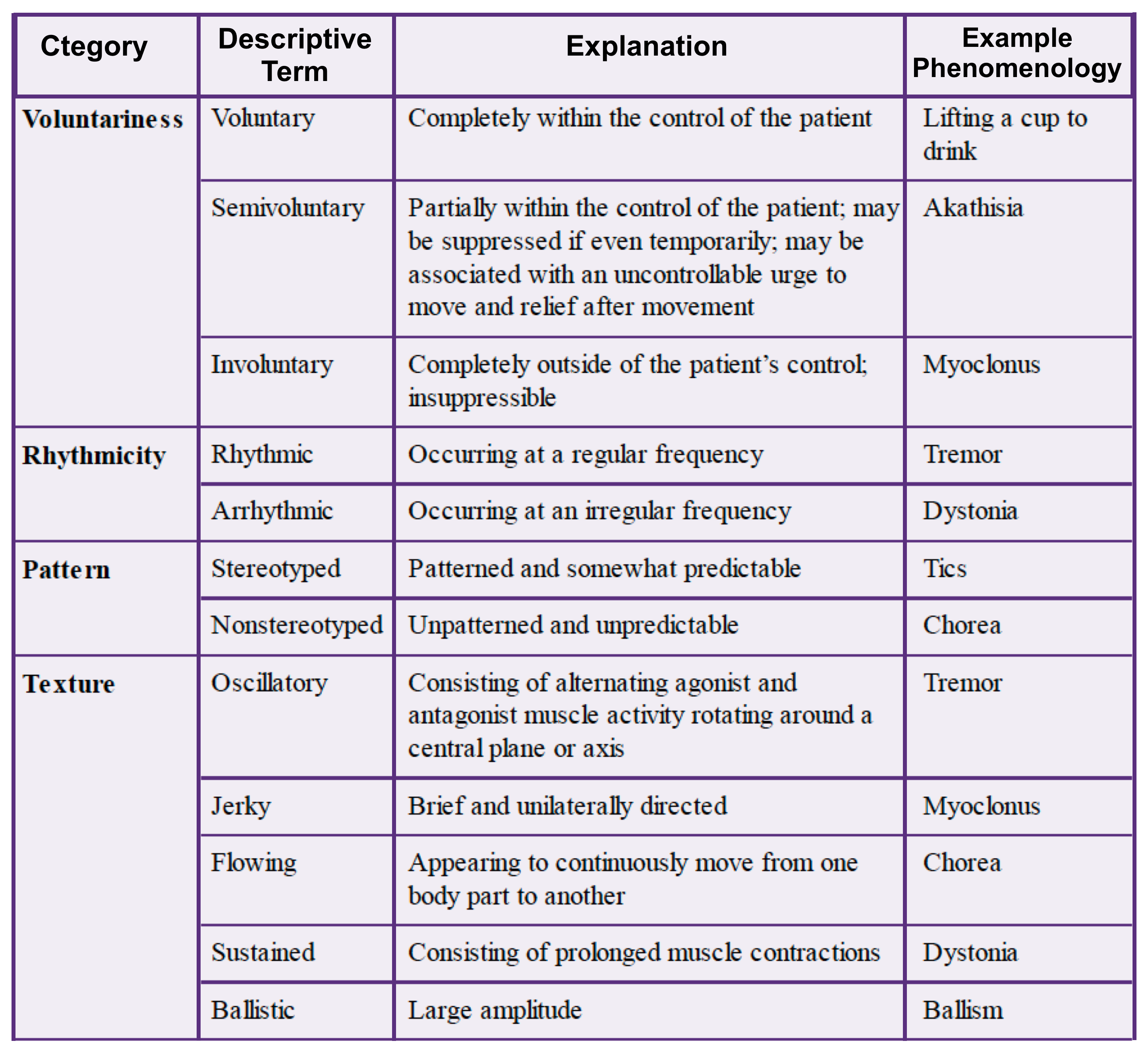
- When describing a hyperkinetic movement, one should start with basic descriptors (e.g. oscillatory, flowing) to determine the correct phenomenology (e.g. tremor, chorea) and reach the appropriate diagnosis (e.g. Huntington’s disease).
- List of hyperkinetic phenomenologies with descriptive terms:
- Dystonia: Involuntary, sustained.
- Chorea: Involuntary, nonstereotyped, flowing
- Ballism: Involuntary, nonstereotyped, ballistic
- Akathisia: Semivoluntary, stereotyped or nonstereotyped, flowing
- Myoclonus: Involuntary, jerky
- Tics: Semivoluntary, stereotyped
- Tremor: Involuntary, rhythmic, oscillatory
In the following discussion, emergency hyperkinetic movement disorders are explored.
Acute Dystonia
- The dystonias refer to a large group of heterogeneous conditions characterized by involuntary sustained or spasmodic muscle contractions involving co-contraction of the agonist and the antagonist’s muscles.
- Movements are usually slow and sustained (unlike chorea) and occur in a repetitive and patterned manner; however, they can be unpredictable and fluctuate.
- A rare life-threatening state of generalized, unremitting severe dystonia (dystonic storm or status dystonicus) is discussed below.
- Acute dystonia can be a presentation of an acute reaction to extrinsic agents (e.g. dopamin blocker agents), or it could be due to a sudden exacerbation of a pre-existing disease (Wilson disease).
- Dystonic reactions are variable in location and severity and are occasionally painful *.
▪️Pathogenesis
- The pathogenesis of acute dystonia may involve dopaminergic hypofunction which leads to a relative overactivity of cholinergic mechanisms *,*.
- This hypothesis is supported by a consistent amelioration of acute dystonia by anticholinergic drugs.
▪️Etiology
- Primary dystonia
- Exacerbation of Parkinson’s disease or related disorders.
- Wilson disease
- Neuroacanthocytosis
- Metabolic disorders: urea cycle disorder
- Secondary dystonia
- Drug-induced dystonia
- Toxic: Carbon monoxide poisoning
- Vascular and structural
- Basal ganglia stroke (especially putaminal lesions).
- Posterior fossa lesions.
- Infection: neurosyphilis, acute herpetic brainstem encephalitis)
- Traumatic brain or peripheral injury
- Inflammatory: Tonic spasms (paroxysmal dystonia) in multiple sclerosis, sarcoidosis
- Degenerative: Laryngeal dystonia in multiple system atrophy
▪️Clinical presentation
- Chronocity
- Acute dystonia usually begins within ~72 hours of medication initiation but may be delayed by as many as 5 days *,*.
- Tardive (late-appearing) dystonia can occur following a minimum of 3 months of neuroleptic treatment *.
- The investigation and acute management of tardive dystonia are largely similar to acute dystonia.
- Clinical features
- Dystonia can be focal, multifocal, segmental, hemidystonia, or generalized.
- Dystonia often affects the face and neck as torticollis, and less commonly the limbs and trunk as opisthotonus *. 🎥
- Oculogyric crisis (OGC) 🎥
- Laryngeal dystonia 🎥
- It may cause airway obstruction with stridor, which can be fatal *.
- The simultaneous presence of other dystonias may help suggest the diagnosis (e.g. blepharospasm, torticollis, or opisthotonos).
- It may cause airway obstruction with stridor, which can be fatal *.
- Orofacial dystonia. 🎥
- Trismus, oromandibular dystonia (e.g. forced jaw opening).
- Tongue protrusion. 🎥
- Blepharospasm. 🎥
- Torticollis. 🎥
▪️Differential diagnosis (pseudodystonias i.e. imitators of dystonia)
- Head tilt (vestibulopathy, trochlear nerve palsy) *
- Atlanto axial and shoulder subluxation
- Arnold-Chiari malformation *
- Neck mass, deep infection, peritonsillar abscess, meningitis *.
- Partial seizure causing abnormal posture.
- Localized tetanus.
- Spasms (hypocalcemia, hypomagnesemia, alkalosis) *
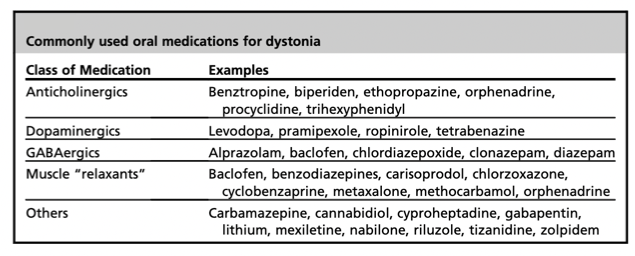
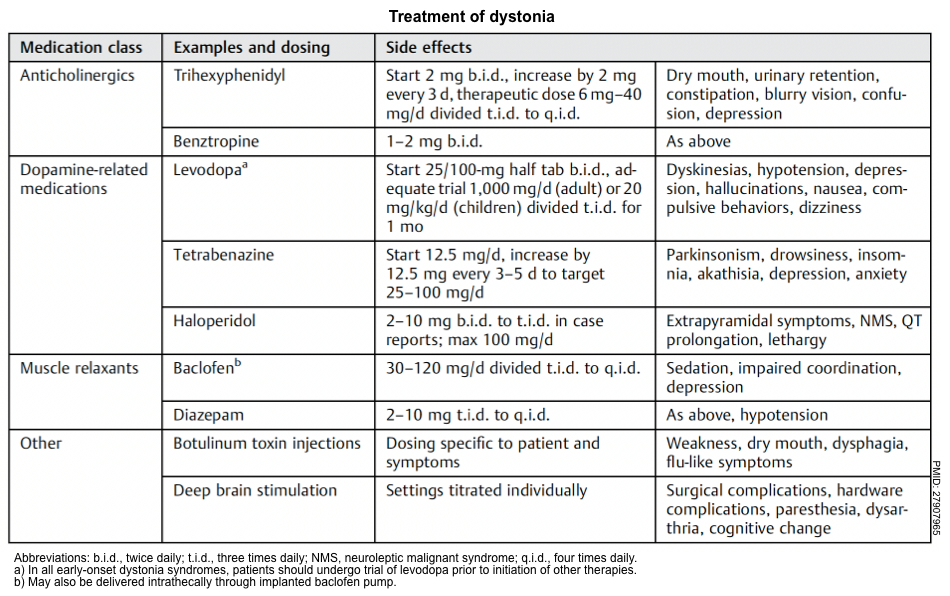
▪️Treatment (symptomatic treatment of dystonia is summarized in the following table).
- Anticholinergic medications are a front-line treatment for dystonia.
- Benztropine
- 💉1-2 mg IV with an additional dose as needed 20 minutes later is often effective.
- 💊An oral course of benztropine tapering off over a week should be used to reduce the risk of recurrence.
- Trihexyphenidyl
- 1 mg/day PO initially; increase as necessary to maintenance range of 5-15 mg/day PO divided q6-8hr *.
- Diphenhydramine
- 💉25-50mg IV is as effective as benztropine and is more widely available *.
- Benztropine
- Benzodiazepines are second-line treatment, only if anticholinergic fails.
- For patients failing therapy, reconsider the possibility of pseudodystonia.
- Levodopa
- For patients with parkinsonism, levodopa is helpful.
- Intubation, if airway obstruction occurs.
- Flexible nasolaryngoscopy should be performed (if possible) before intubation to secure the diagnosis (to exclude other forms of anatomic obstruction that could cause stridor).
- Rapid sequence intubation with full paralysis may facilitate safe intubation.
- 👉Note that if sedation-only intubation is attempted, dystonia may impair attempts at intubation (e.g. persistent vocal cord adduction may not allow passage of the endotracheal tube).
- Patients with torticollis may have cervical spine instability, so caution should be taken during airway manipulation.
▪️Overview of treatment of dystonia
Dystonic storm
- Dystonic storm or status dystonicus is a rare life-threatening state of generalized, unremitting severe dystonic spasm.
- This is a malignant hyperkinetic movement disorder emergency in which rapid deterioration of dystonia requires emergent intervention *.
- A dystonic storm is frequently refractory to standard antidystonia oral medications.
- Mortality is high (~10%) even with treatment *.
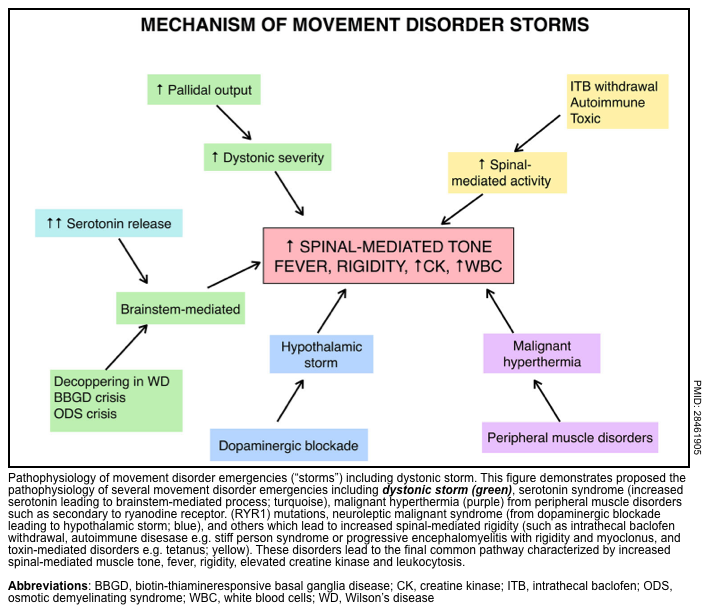
▪️Pathophysiology
- In patients with underlying dystonia, increased dystonia severity may result from increased pallidal output. A hypothetical framework for conceptualizing possible mechanisms of various storm conditions is represented below.
▪️Etiology
- It usually emerges gradually after weeks or months in the patient with an underlying dystonia diagnosis.
- However, status dystonicus may also present during new onset dystonic disorders without previous or only mild dystonic movements *.
- Underlying causes of dystonia include *
- Genetic dystonias.
- Cerebral palsy.
- Post-traumatic dystonia.
- Wilson’s disease.
- Infectious or autoimmune encephalitis (e.g. anti-NMDA-receptor encephalitis).
▪️Triggers of dystonic storms
- Infections are the most common triggers (particularly gastroenteritis with dehydration) *
- Medication changes.
- Hyperthermia, dehydration, metabolic abnormalities *.
- Trauma/surgery.
- Penicillamine therapy for Wilson’s disease (generally with initiation of therapy) *.
- Failure of a deep brain stimulator device.
▪️Clinical presentation
- Clinical course
- Dystonic storm usually occurs in patients who are already known to have dystonia.
- It tends to occur in patients with severe or poorly controlled dystonia at baseline.
- In some patients a prodrome of a dystonic storm may occur, where dystonia is worsened from baseline but not as severe as in a true storm (video below).
- Once a true storm begins, it usually lasts 2–4 weeks with gradual recovery.
- Dystonic storm usually occurs in patients who are already known to have dystonia.
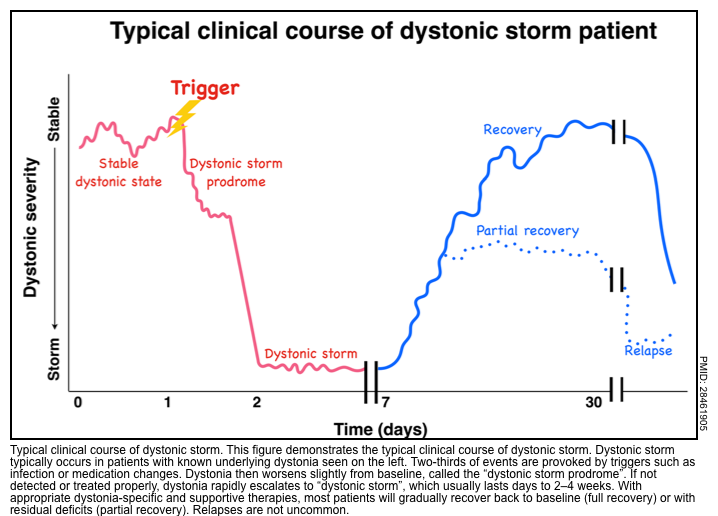
- Clinical features
- Dystonia
- Dystonia is often generalized and sustained.
- Dystonia can be tonic (i.e. sustained posturing), or phasic (i.e. irregular jerking) which is rapid and repetitive *.
- Symptoms and signs of the dystonic storm (these signs & symptoms may help to diagnose a dystonic storm):
- 💡Fever, tachycardia, tachypnea or respiratory change, hypertension, sweating, and autonomic instability*.
- Other movement disorders such as chorea, ballism, or myoclonus may accompany dystonia *.
- Bulbar impairments such as dysarthria, dysphagia, and respiratory failure are common *.
- Pain (requires aggressive symptomatic control) *.
- Dystonia
- Laboratory findings
- Leukocytosis, ⬆️serum CK, ⬆️CRP, myoglobinemia, and myoglobinuria that may be severe enough to lead to renal failure and academia *.
- Complications
- The unremitting dystonic spasms may lead to hyperpyrexia, dehydration, respiratory failure, and rhabdomyolysis with renal failure.*
▪️Differential diagnosis
- Considerations
- Several conditions may mimic dystonic storm, presenting with hyperthermia, elevated serum CK, and/or muscle rigidity.
- It is important to differentiate dystonic storms from other hyperkinetic movement disorder emergencies.
- The phenomenology of the underlying movement disorder, associated neurological symptoms and signs, age of the patient, history of triggers, and time course are helpful clues.
- Compared to other movement disorder emergencies, dystonic storm typically occurs in the pediatric age group. Triggers may or may not be present, and a storm usually develops quickly over hours to several days.
- Differentials (see following table *)
- Neuroleptic malignant syndrome, serotonin syndrome, malignant hyperthermia.
- Acute dystonic drug reaction.
- Status epilepticus.
- Paroxysmal autonomic instability with dystonia.
- Intrathecal baclofen withdrawal.
- Acute parkinsonism *.
- Autoimmune encephalitis e.g. Anti-NMDA (video below)
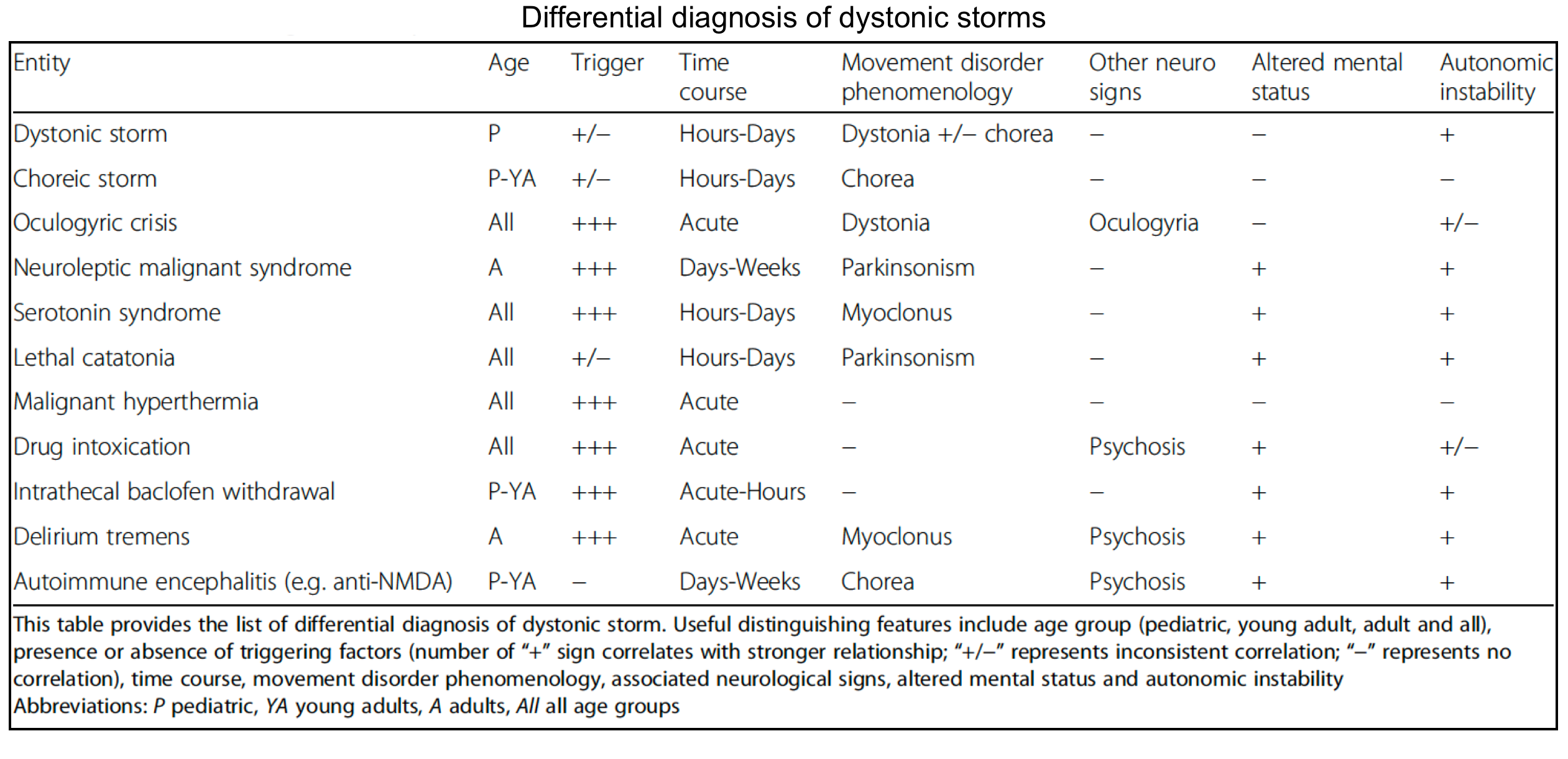
▪️Management
- Supportive care
- Any recently withdrawn medications should be restarted.
- Treat the underlying precipitant.
- Provide supportive care:
- Antipyretics
- IV Antibiotics
- Nutritional supports
- Management of rhabdomyolysis 📖
- Multimodal pain management 📖
- Sedation: Clonidine or in more severe cases benzodiazepine. For intubated patients, propofol is most often used.
- Specific treatment
- Non-invasive
- Anticholinergic medications (e.g. trihexyphenidyl, benztropine).
- Baclofen.
- Benzodiazepines.
- Clonidine.
- Tetrabenazine (catecholamine depleter)
- Invasive therapies
- Intrathecal baclofen
- Deep brain stimulation
- Non-invasive
- Intensive care
- Intubation and continuous sedation
- A review of 37 cases of dystonic storm * reported that patients benefitted from escalation to intensive care units for endotracheal intubation, mechanical ventilation, sedation with IV midazolam, and propofol. Early escalation to intensive care is therefore suggested *.
- In medication-refractory cases, intubation and sedation may be necessary (e.g. with propofol). For the most refractory cases, paralysis is the last resort for initial stabilization *.
- Recovery may take days or even weeks. In refractory cases, intrathecal baclofen or bilateral deep-brain stimulation may accelerate recovery
- Intubation and continuous sedation
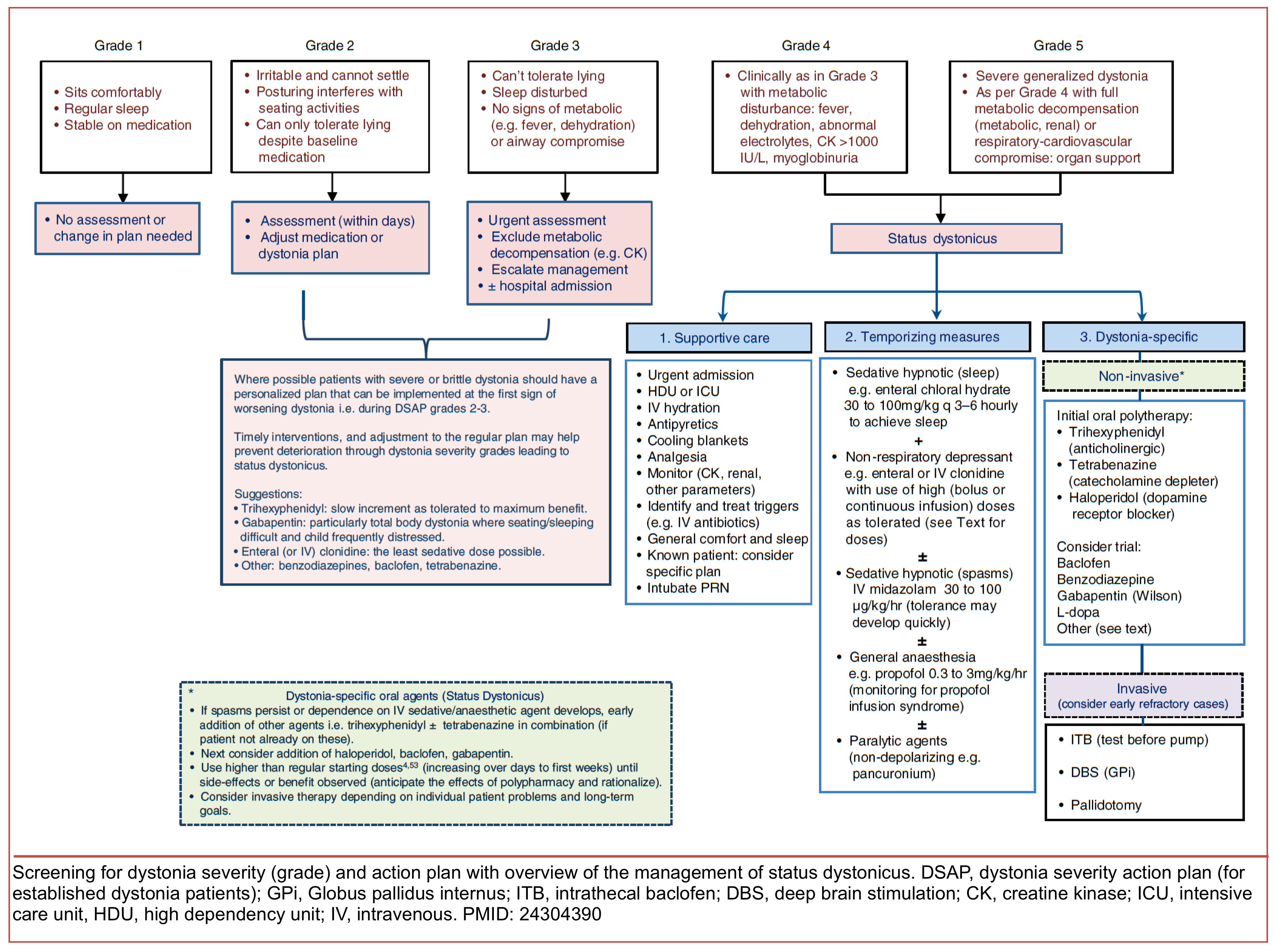
Akathisia
Akathisia refers to the subjective report of inner restlessness particularly referable to the legs, with a consequent inability to maintain a posture for several minutes, and the objective manifestations of restlessness in the form of movements of the limbs, a tendency to shift body position in the chair while sitting or marching while standing *. 🎥
▪️Etiology
- Akathisia usually starts within hours or days after the initiation or increase in antidopaminergic agents (e.g. antipsychotics) dosage or change to a high potency antidopaminergic agent *.
▪️Differential diagnosis
- Delirium
- The inability to stay still may be misinterpreted as representing agitation due to delirium (which could lead to a vicious spiral of increased antipsychotic dosing) *.
- Restless legs syndrome (RLS)
- RLS can be interpreted as focal akathisia mostly affecting legs but with a clear circadian pattern, being worse in the evening *.
▪️Management
- Withdrawal of the offending medication.
- Supportive treatment to relieve distressing symptoms:
- Anticholinergics (see above)
- Use the lowest effective dose and monitor for peripheral anticholinergic side effects (e.g. constipation, dry mouth) and central anticholinergic side effects (confusion, memory disturbance), especially in the elderly *.
- Antiadrenergic such as propranolol or clonidine *.
- Serotonin antagonists such as mirtazapine * or trazodone *.
- Anticholinergics (see above)
Chorea, and Ballism
- Chorea, athetosis, and ballism share a common underlying pathology. Although they are often referred to separately, the distinction between chorea, athetosis, and ballism are somewhat arbitrary and relate more to the phenomenology (speed, amplitude, and duration of the movement) rather than underlying pathology *.
- Chorea is defined as a movement characterized by abrupt continuous, nonrhythmic, nonpatterned involuntary movements.
- Athetosis (aka choreoathetosis) identifies a slow and distal form of chorea that, because of the slowness of the movement, has a twisting or snakelike appearance.
- Ballism can be considered to be a very severe form of chorea in which the movements are proximal and have violent, flinging/flailing movements of an extremity.
- Hemiballismus or hemichorea refers to these movements involving half of the body, which is the most common distribution. 🎥, 🎥
- Chorea is defined as a movement characterized by abrupt continuous, nonrhythmic, nonpatterned involuntary movements.
- Any ischemic, infectious, inflammatory, hemorrhagic, or mass lesion that affects the caudate or subthalamic nucleus may cause chorea or hemiballismus, respectively.
▪️Etiology
- Toxins
- Carbon monoxide, manganese, thallium, toluene, methanol, cyanide, mercury, cocaine, heroin, amphetamines.
- Metabolic
- Hyper/hypoglycemia especially diabetic ketoacidosis or hyperglycemic hyperosmolar state (HHS)
- Hyper/hyponatremia
- Hypomagnesemia
- Hypocalcemia
- Thyrotoxicosis
- Chronic acquired hepatocerebral degeneration
- Uremic encephalopathy.
- Wilson’s disease.
- Polycythemia vera.
- Drug-induced
- Autoimmune/inflammatory
- Antiphospholipid antibody syndrome.
- Systemic lupus erythematosus.
- Behcet disease.
- Sydenham’s chorea (post beta-hemolytic streptococcal infection).
- Although the diagnosis is clinical, elevated antistreptolysin-O antibody titers usually secure the diagnosis.
- Chorea gravidarum (usually beginning within the first 12 weeks of pregnancy) *.
- Anti-NMDA (N-methyl D-aspartate) receptor encephalitis.
- Infections
- Creutzfeldt–Jakob disease
- HIV
- Toxoplasmosis
- Tuberculoma
- Central nervous system (Structural lesions of the subthalamic nucleus0
- Vascular: Hemorrhagic or ischemic lesions
- Space-occupying lesions (e.g. tumors, vascular malformations).
- Multiple sclerosis.
▪️Management
- Identify and treat the underlying pathology e.g. hyperglycemic hyperosmolar state, infection, metabolic abnormality.
- Spontaneous resolution generally occurs eventually (within three months), so treatment is not necessary if the symptoms are manageable.
- Specific treatment
- If treatment is required (e.g. violent, self-injurious, exhausting, or distressing movements), chorea can be symptomatically treated with many classes of medications:
-
- Dopamine antagonists such as neuroleptics, reserpine, and tetrabenazine.
- Atypical antipsychotics such as clozapine, quetiapine, or risperidone.
- Antiseizure medications such as benzodiazepines, valproate, carbamazepine, or levetiracetam
- N-methyl-D-aspartate- (NMDA) receptor antagonists such as amantadine or riluzole.
- Dopamine antagonists such as neuroleptics, reserpine, and tetrabenazine.
-
- If treatment is required (e.g. violent, self-injurious, exhausting, or distressing movements), chorea can be symptomatically treated with many classes of medications:
Myoclonus
- Myoclonus is defined as a sudden, brief, shock‑like, involuntary movement caused by muscular contractions.
- Myoclonus can sometimes happen as a transient loss of muscle tone (referred to as a form of negative myoclonus). This is best seen as asterixis.
- Different forms of myoclonus exist. They can be classified according to their distribution, relationship to precipitating stimuli, and site of origin (table below) or etiology. *
- Based on body distribution, myoclonus is classified as focal, multifocal, segmental, or generalized.
- Generalized myoclonus has a widespread distribution, whereas focal or segmental myoclonus is restricted to a particular part of the body.
- In segmental myoclonus, the generator in the brainstem or spinal cord produces movements at that particular segment or a few close contiguous segments, giving the jerks a focal or circumscribed distribution.
- It is unaffected by supraspinal influences like state of consciousness (it persists in sleep), voluntary action (therefore present at rest), and sensory
stimulus. - Palatal myoclonus (🎥) is the most common type followed by spinal segmental myoclonus.
- It is unaffected by supraspinal influences like state of consciousness (it persists in sleep), voluntary action (therefore present at rest), and sensory
- Based on relation to activity, it can be differentiated as myoclonus occurring at rest, during an action, or maintaining a posture.
- Myoclonus can be spontaneous, or it can be brought on by sensory stimulation, arousal, or the initiation of movement (action myoclonus, aka. reflex or stimulus-sensitive myoclonus).
- Physiological myoclonus
- It refers to myoclonic jerks occurring in healthy people. It produces minimal or no disability, and examination is usually normal. The examples of physiological myoclonus are as follows:
- Nocturnal myoclonus
- Hiccup
- Benign infantile myoclonus with feeding
- It refers to myoclonic jerks occurring in healthy people. It produces minimal or no disability, and examination is usually normal. The examples of physiological myoclonus are as follows:
- Based on body distribution, myoclonus is classified as focal, multifocal, segmental, or generalized.
▪️Site of origin
- Cortical myoclonus
- It usually tends to be either focal or multifocal but can be generalized, and it is particularly aggravated by action and tactile stimuli.
- Cortical‑subcortical myoclonus
- Typically seen in generalized epilepsy syndromes, is usually generalized or multifocal and occurs at rest. However, it can be stimulus-sensitive.
- Brainstem reticular reflex myoclonus (generated from the lower brainstem reticular formation in the medulla)
- It presents with generalized, synchronously involving both the arms proximally as well as the neck, face, trunk, and lower limbs.
- These jerks are more flexor than extensor, occur at rest and during action, and are induced by auditory or tactile stimuli such as tapping on the limbs or face.
- Spinal myoclonus
- It presents with segmental, unilateral arrhythmic jerking in the arm and/or trunk.
- Propriospinal myoclonus arises in the spinal cord and then spreads up and down the cord, leading to a brief bodily contraction.
- Peripheral myoclonus (due to a lesion in the nerve, plexus, or root)
▪️Causes of myoclonus *
- Epileptic myoclonus (a component of a seizure disorder):
- Focal motor seizures (including epilepsia partialis continua).
- Myoclonic epilepsy.
- Myoclonic status epilepticus (MSE) occurs in patients with global anoxic brain injuries and has been historically felt to be a poor prognostic sign.
- Metabolic disorders
- Hypoglycemia/ Hyperosmolar nonketotic hyperglycemia
- Hyponatremia/ Hypernatremia
- Hypomagnesemia.
- Hepatic encephalopathy
- Uremia, dialysis syndrome
- Hypoxia (Lance–Adams syndrome)
- Hyperthyroidism.
- Vitamin B12 deficiency, Vitamin E deficiency, Wernicke encephalopathy.
- Medications
- Antipsychotics (including tardive myoclonus from chronic exposure).
- Antidepressants (TCA, SSRIs, SNRIs, monoamine oxidase inhibitors): Serotonin syndrome
- Lithium.
- Parkinson’s medications (levodopa, bromocriptine, amantadine, selegiline).
- Antiepileptics (phenytoin, valproate, carbamazepine, lamotrigine, vigabatrin, phenobarbital, primidone).
- Withdrawal of medications (e.g. alcohol, benzodiazepine, or antiepileptic agents).
- Sympathomimetic intoxication.
- Antibiotics (e.g. penicillins; cephalosporins, especially cefepime; carbapenems; quinolones).
- Midazolam, propofol.
- Opioids (morphine, fentanyl, tramadol)
- Other:
- Lidocaine.
- Gabapentin.
- Antiarrhythmics (e.g., amiodarone, flecainide).
- Diltiazem, verapamil.
- Anticancer drugs (chlorambucil, busulfan, cyclophosphamide, ifosfamide).
- Contrast media
- Degenerative disorders
- Wilson disease
- Huntington disease
- Myoclonus dystonia
- Alzheimer disease
- Parkinson’s disease and Parkinson-plus disorders
- Spinocerebellar degenerations
- Focal brain or nerve damage
- Head injury
- Stroke
- Tumors
- Peripheral nerve or plexus injury (physical injury, tumor); causing focal myoclonus.
- Inflammation (e.g. multiple sclerosis).
- Dementia
- Alzheimer’s disease.
- Creutzfeldt-Jakob disease.
- Dementia with Lewy bodies.
- Frontotemporal dementia, corticobasal degeneration.
- Inflammation (e.g. infectious, postinfectious, paraneoplastic):
- Viral encephalitis (arbovirus, HSV), postinfectious encephalitis.
- Lyme, HIV, malaria, syphilis, cryptococcus, progressive multifocal leukoencephalopathy.
- Anti-NMDA-receptor encephalitis, paraneoplastic encephalopathies.
- Hashimoto encephalopathy.
▪️Differential diagnosis
- Psychogenic myoclonus. 🎥
- Functional or psychogenic involuntary movements are highly variable, manifesting as shaking, fixed postures, bizarre gaits, paroxysmal disorders, or other phenomena (table below).
- Characteristic features of psychogenic myoclonus include sudden onset and remission, constantly changing anatomical and temporal patterns, distractibility, variable amplitude, inconsistency over time, and placebo response.
- This is aggravated by stress and anxiety, and there is exaggerated stimulus sensitivity to visual or auditory stimuli.
- Chorea, dystonia, tremor
▪️Evaluation
- Review of medication list & drug interactions.
- Electrolytes (including calcium and magnesium).
- Glucose.
- Liver function tests.
- Thyroid function tests.
- EEG to exclude seizure.
- Neuroimaging.
- Workup for infection, if clinically indicated.
▪️Treatment
- Identify and treat the possible underlying causes (e.g., removal of offending drug/s or toxin/s, correction of metabolic or electrolyte disturbances).
- Treatments available for persistent and debilitating myoclonus include (a combination of medications are often necessary) *:
- Clonazepam
- Piracetam
- Valproic acid
- Levetiracetam *
- Postanoxic action myoclonus is remarkably responsive to 5-hydroxytryptophan, the precursor of the neurotransmitter 5-hydroxytryptamine (serotonin).
- The 5-hydroxytryptophan is increased gradually to a maximum of 1 to 1.5 mg/d orally and may be combined with carbidopa (maximum, 400 mg/d orally) to inhibit metabolism in peripheral tissues.
- Some antiseizure drugs, such as phenytoin, may worsen myoclonus and should be avoided if possible *.
Posthypoxic myoclonus
▪️General
- Posthypoxic myoclonus (PHM) is a complication occurring in 18–37% of patients who enter a coma after cardiopulmonary resuscitation (CPR) from different causes, such as cardiac arrest, surgical complications, respiratory arrest due to asthma attacks, anesthesia, or drug intoxication.
- Two types of posthypoxic myoclonus exist:
- Acute PHM (myoclonus status epilepticus)
- Chronic form of PHM (Lance-Adams syndrome, LAS)
- Differentiation between these two entities is therefore critically important because they have different prognostic implications (table below).
▪️Acute PHM (aka myoclonic status epilepticus) *
- Onset within <48 hours of arrest.
- Generalized myoclonus involves four limbs and the face. This should be synchronized, repetitive, symmetric, and unequivocal (not just a little jerking of the hands or face).
- Persists for >30 minutes.
- Associated with clinical coma.
- EEG shows either status epilepticus or a malignant pattern (e.g. burst suppression) *.
- Poor survival (~10%).
▪️Chronic form of PHM (Lance-Adams syndrome, LAS)
- Myoclonus emerges later after the occurrence of cardiac arrest, often after several days (after regaining consciousness).
- Patients are typically awake.
- Myoclonus occurs while the patient is attempting to move (intention myoclonus), or may be triggered by stimulation.
- This tends to be persistent.
- Lance-Adams syndrome does not carry the same negative prognostic implications as status myoclonus. About half of patients with Lance-Adams syndrome may have a slow neurological recovery.
▪️Management
- Acute PHM
- Antiepileptic medications are usually first-line treatments, especially when seizures coexist
- Benzodiazepines, phenytoin, phenobarbital, and valproic acid have been used in this setting following the dosages normally used for convulsive status epilepticus.
- Phenytoin should be used with caution since it can paradoxically worsen subcortical myoclonus.
- Propofol has been utilized to control the symptoms if generalized myoclonus does not respond to antiepileptics.
- Chronic PHM
- First-line treatments consist of antiepileptic drugs, especially clonazepam (up to 15 mg/day), levetiracetam (up to 3000 mg/day), and valproic acid (up to 2000 mg/day).
- Other medications have been attempted in patients with PHM including intrathecal baclofen, lacosamide, levodopa, carbamazepine, chlorpromazine, paroxetine, estrogen, agomelatine, zonisamide, and cannabidiol.
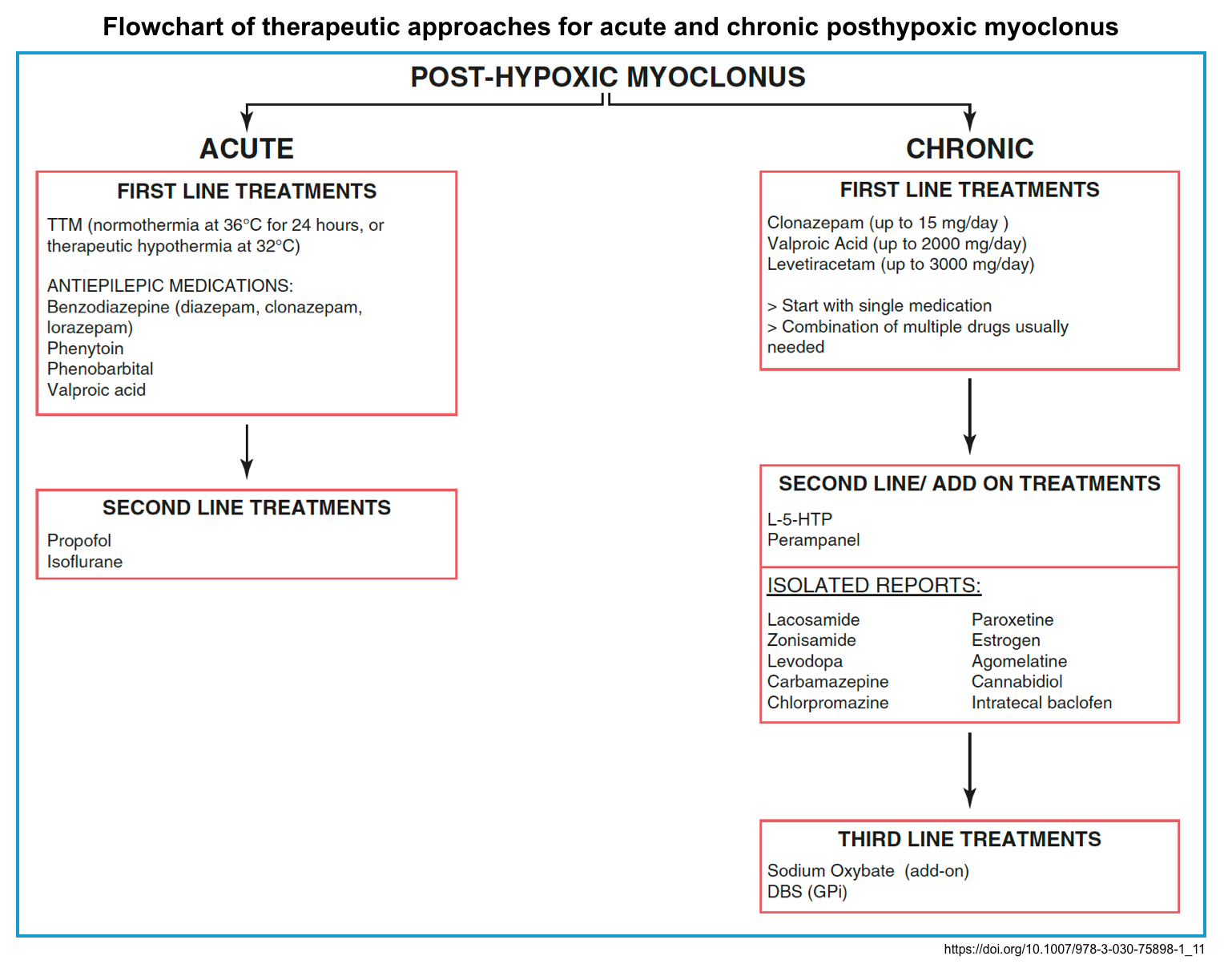
Tetanus
▪️General
- Tetanus results from intoxication with Clostridium tetani.
- An exotoxin produced by the Clostridium species (aka. tetanospasmin) enters peripheral nerves, is transported to the alpha motor neurons via retrograde axonal transport, and antagonizes GABA and glycine release (inhibitory transmitters), leading to profound autonomic dysregulation and sustained motor neuron activity with muscle spasm *.
▪️Clinical features
- Course
- The incubation period following inoculation into the soft tissue may range from a few days to several weeks.
- Prodromal symptoms may include:
- Headache.
- Trismus (painless lockjaw) is seen in most cases. This may be associated with neck stiffness and difficulty swallowing.
- Fasciculations may occur at the wound site.
- Full-blown clinical presentation
- Severe, unopposed muscle contractions that may be generalized or local (such as in the face), autonomic overactivity, and sometimes seizures.
- Patients may display trismus (lock-jaw) 🎥, risus sardonicus (spasm of facial muscles), and/or opisthotonus (dramatic extension of the neck, back, and legs) 🎥. *
- Involvement of pharyngeal and/or thoracic muscles may cause respiratory arrest.
- Autonomic features include hyperpyrexia, hypertension, tachycardia, diaphoresis, and urinary retention *.
- Patients may display trismus (lock-jaw) 🎥, risus sardonicus (spasm of facial muscles), and/or opisthotonus (dramatic extension of the neck, back, and legs) 🎥. *
- Severe, unopposed muscle contractions that may be generalized or local (such as in the face), autonomic overactivity, and sometimes seizures.
▪️Diagnosis and differentials
- Tetanus is diagnosed clinically, as it is difficult to isolate the responsible bacterium from wounds or blood.
- Other differential diagnostic possibilities that should be considered include:
- Neuroleptic malignant syndrome.
- Drug-induced dystonic reactions *.
- Trismus due to a dental infection.
- Strychnine poisoning due to rat poison ingestion.
- Stiff-person syndrome (SPS)
- Hypocalcemia.
▪️Management
- Control of muscle spasms
- Benzodiazepines: High doses are used to control spasms *.
- Neuromuscular blockade
- In extreme cases, intubation with deep sedation or even paralysis may be required.
- Magnesium sulfate
- Magnesium is a presynaptic neuromuscular blocker, that blocks catecholamine release from nerves, and reduces receptor responsiveness to catecholamines *.
- Magnesium sulfate infusion has been shown to reduce the requirement for other medications to manage muscle spasms and cardiovascular instability *.
- Management of autonomic dysfunction
- Several drugs can be used to produce adrenergic blockade and suppress autonomic hyperactivity. These include:
- Magnesium sulfate infusion: Loading dose 40 mg/kg over 30 minutes, followed by continuous infusion of either 2 g per hour for patients over 45 kg
or 1.5 g per hour for patients ≤45 kg. - Esmolol
- Morphine sulfate: 0.5 to 1 mg/kg per hour by continuous intravenous infusion is commonly used to control autonomic dysfunction as well as to induce sedation.
- Magnesium sulfate infusion: Loading dose 40 mg/kg over 30 minutes, followed by continuous infusion of either 2 g per hour for patients over 45 kg
- Several drugs can be used to produce adrenergic blockade and suppress autonomic hyperactivity. These include:
- Reducing toxin production
- Wound management
- All patients with tetanus should undergo wound debridement to eradicate spores and necrotic tissue, which could lead to conditions ideal for germination *.
- Antibiotic therapy
- Wound management
- Neutralization of unbound toxin
- Since tetanus toxin is irreversibly bound to tissues, only unbound toxin is available for neutralization.
- Tetanus immunoglobulin is administered intramuscularly or intravenously; this removes unbound tetanus toxin but does not affect toxin that is already bound to nerve endings *.
- Active immunization
- Since tetanus is one of the few bacterial diseases that do not confer immunity following recovery from acute illness, all patients with tetanus should receive
active immunization with a full series (e.g. three doses in adults and children >7 years old) of tetanus and diphtheria toxoid-containing vaccines, commencing immediately upon diagnosis. Such vaccines should be administered at a different site than tetanus immune globulin.
- Since tetanus is one of the few bacterial diseases that do not confer immunity following recovery from acute illness, all patients with tetanus should receive
Appendix
Receptor-binding profile of antipsychotic medications
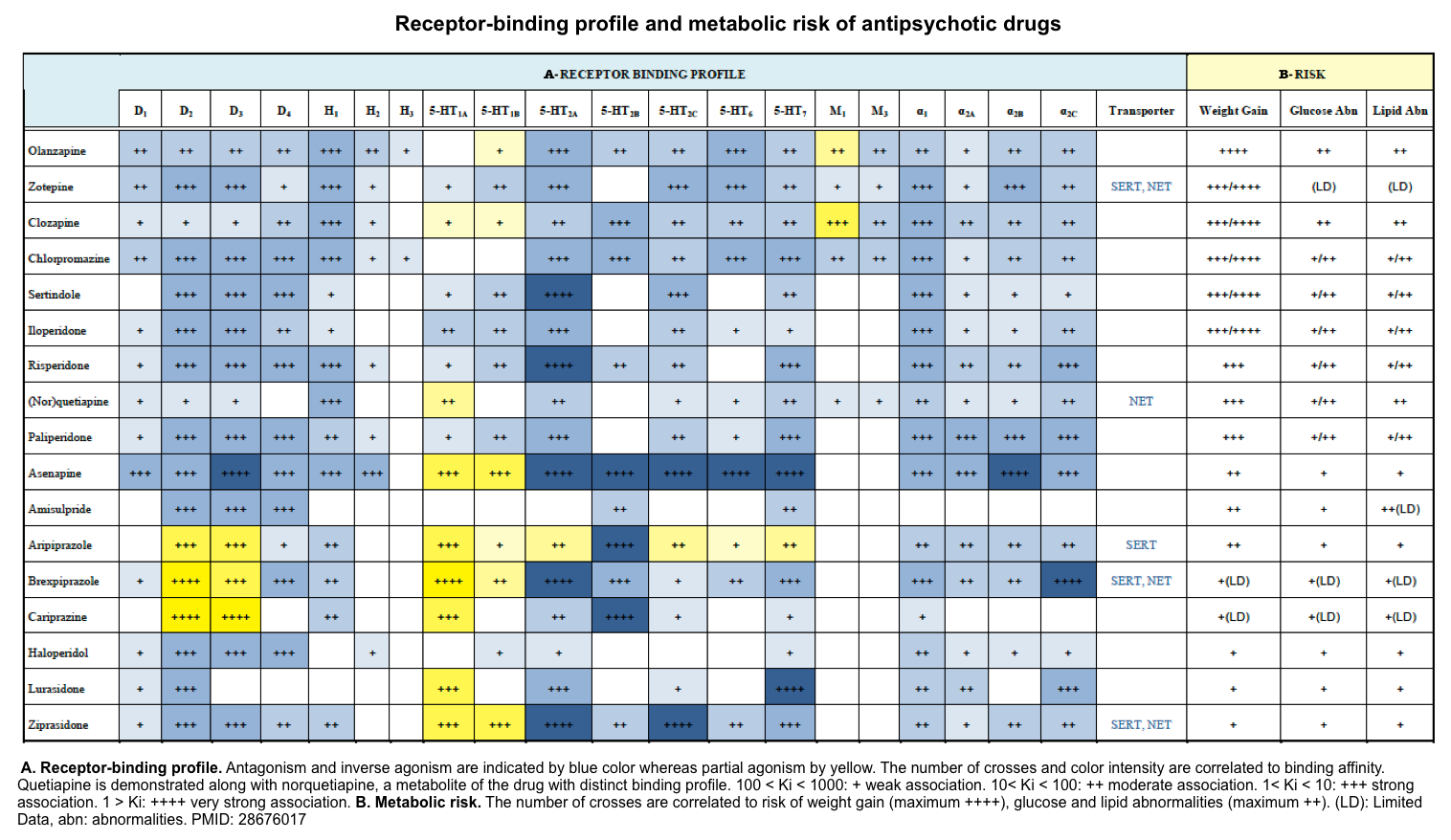
Selected adverse effects of antipsychotic medications
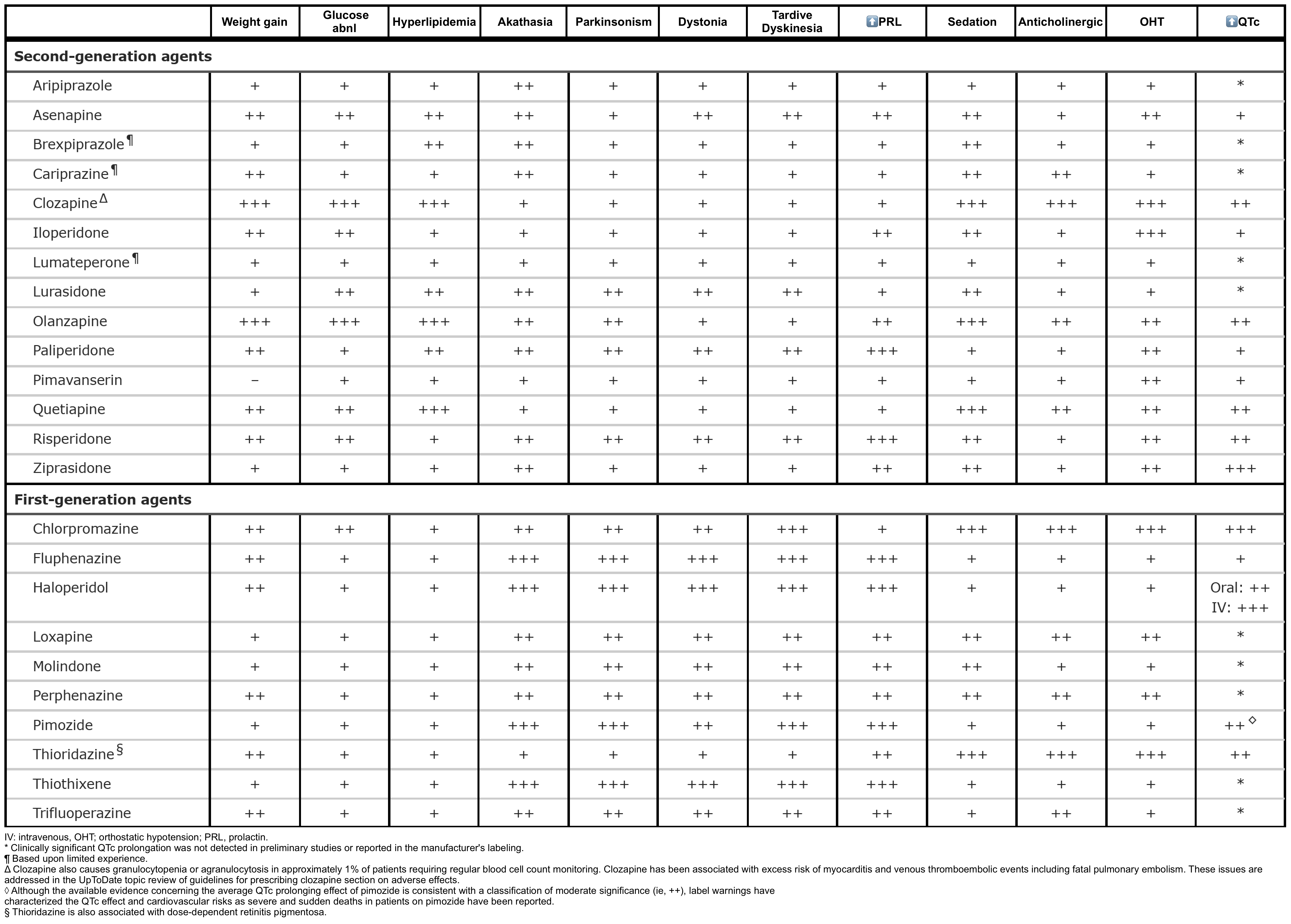
Drugs used in the treatment of Alzheimer’s disease
References
1. PMID: 27907965. Schaefer SM, Rostami R, Greer DM. Movement Disorders in the Intensive Care Unit. Semin Neurol. 2016 Dec;36(6):607-614. doi: 10.1055/s-0036-1592356. Epub 2016 Dec 1.
2. PMID: 32659853. Kuhlwilm L, Schönfeldt-Lecuona C, Gahr M, Connemann BJ, Keller F, Sartorius A. The neuroleptic malignant syndrome-a systematic case series analysis focusing on therapy regimes and outcome. Acta Psychiatr Scand. 2020 Sep;142(3):233-241. doi: 10.1111/acps.13215. Epub 2020 Aug 2.
3. PMID: 17541044. Strawn JR, Keck PE Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007 Jun;164(6):870-6. doi: 10.1176/ajp.2007.164.6.870.
4. PMID: 23194104. Langan J, Martin D, Shajahan P, Smith DJ. Antipsychotic dose escalation as a trigger for neuroleptic malignant syndrome (NMS): literature review and case series report. BMC Psychiatry. 2012 Nov 29;12:214. doi: 10.1186/1471-244X-12-214.
5. PMID: 28144147. Oruch R, Pryme IF, Engelsen BA, Lund A. Neuroleptic malignant syndrome: an easily overlooked neurologic emergency. Neuropsychiatr Dis Treat. 2017 Jan 16;13:161-175. doi: 10.2147/NDT.S118438.
6. PMID: 12151905. Gurrera RJ. Is neuroleptic malignant syndrome a neurogenic form of malignant hyperthermia? Clin Neuropharmacol. 2002 Jul-Aug;25(4):183-93. doi: 10.1097/00002826-200207000-00001.
7. PMID: 27423483. Pileggi DJ, Cook AM. Neuroleptic Malignant Syndrome. Ann Pharmacother. 2016 Nov;50(11):973-981. doi: 10.1177/1060028016657553. Epub 2016 Jul 19.
8. PMID: 26110802. Munhoz RP, Scorr LM, Factor SA. Movement disorders emergencies. Curr Opin Neurol. 2015 Aug;28(4):406-12. doi: 10.1097/WCO.0000000000000212.
9. PMID: 30459031. Tormoehlen LM, Rusyniak DE. Neuroleptic malignant syndrome and serotonin syndrome. Handb Clin Neurol. 2018;157:663-675. doi: 10.1016/B978-0-444-64074-1.00039-2.
10. PMID: 28811916. Sarkar S, Gupta N. Drug information update. Atypical antipsychotics and neuroleptic malignant syndrome: nuances and pragmatics of the association. BJPsych Bull. 2017 Aug;41(4):211-216. doi: 10.1192/pb.bp.116.053736.
11. PMID: 28461905. Termsarasab P, Frucht SJ. Dystonic storm: a practical clinical and video review. J Clin Mov Disord. 2017 Apr 28;4:10. doi: 10.1186/s40734-017-0057-z.
12. PMID: 30743298. Rajan S, Kaas B, Moukheiber E. Movement Disorders Emergencies. Semin Neurol. 2019 Feb;39(1):125-136. doi: 10.1055/s-0038-1677050. Epub 2019 Feb 11.
13. PMID: 31523132. Scotton WJ, Hill LJ, Williams AC, Barnes NM. Serotonin Syndrome: Pathophysiology, Clinical Features, Management, and Potential Future Directions. Int J Tryptophan Res. 2019 Sep 9;12:1178646919873925. doi: 10.1177/1178646919873925.
13. PMID: 33456910. Bartakke A, Corredor C, van Rensburg A. Serotonin syndrome in the perioperative period. BJA Educ. 2020 Jan;20(1):10-17. doi: 10.1016/j.bjae.2019.10.003. Epub 2019 Dec 4. Erratum in: BJA Educ. 2020 Apr;20(4):139.
14. PMID: 32305961. Yang L, Tautz T, Zhang S, Fomina A, Liu H. The current status of malignant hyperthermia. J Biomed Res. 2019 May 30;34(2):75-85. doi: 10.7555/JBR.33.20180089.
15. PMID: 14661655. Ali SZ, Taguchi A, Rosenberg H. Malignant hyperthermia. Best Pract Res Clin Anaesthesiol. 2003 Dec;17(4):519-33. doi: 10.1016/j.bpa.2003.09.012.
16. PMID: 32008650. Ellinas H, Albrecht MA. Malignant Hyperthermia Update. Anesthesiol Clin. 2020 Mar;38(1):165-181. doi: 10.1016/j.anclin.2019.10.010.
17. PMID: 27907965. Schaefer SM, Rostami R, Greer DM. Movement Disorders in the Intensive Care Unit. Semin Neurol. 2016 Dec;36(6):607-614. doi: 10.1055/s-0036-1592356. Epub 2016 Dec 1.
18. PMID: 19884605. Fink M, Taylor MA. The catatonia syndrome: forgotten but not gone. Arch Gen Psychiatry. 2009 Nov;66(11):1173-7. doi: 10.1001/archgenpsychiatry.2009.141.
19. PMID: 18070843. Carroll BT, Goforth HW, Thomas C, Ahuja N, McDaniel WW, Kraus MF, Spiegel DR, Franco KN, Pozuelo L, Muñoz C. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007 Fall;19(4):406-12. doi: 10.1176/jnp.2007.19.4.406.
20. PMID: 33896535. Apetauerova D, Patel PA, Burns JD, Lerner DP. Movement Disorder Emergencies. Neurol Clin. 2021 May;39(2):615-630. doi: 10.1016/j.ncl.2021.01.005. Epub 2021 Mar 31.
21. PMID: 32067538. Mormando C, Francis A. Catatonia revived: a unique syndrome updated. Int Rev Psychiatry. 2020 Aug-Sep;32(5-6):403-411. doi: 10.1080/09540261.2020.1723500. Epub 2020 Feb 18.
22. PMID: 28917389. Beach SR, Gomez-Bernal F, Huffman JC, Fricchione GL. Alternative treatment strategies for catatonia: A systematic review. Gen Hosp Psychiatry. 2017 Sep;48:1-19. doi: 10.1016/j.genhosppsych.2017.06.011. Epub 2017 Jun 24.
23. PMID: 21555633. Robottom BJ, Weiner WJ, Factor SA. Movement disorders emergencies. Part 1: Hypokinetic disorders. Arch Neurol. 2011 May;68(5):567-72. doi: 10.1001/archneurol.2011.84.
24. PMID: 23449883. Siniscalchi A, Gallelli L, Labate A, Malferrari G, Palleria C, Sarro GD. Post-stroke Movement Disorders: Clinical Manifestations and Pharmacological Management. Curr Neuropharmacol. 2012 Sep;10(3):254-62. doi: 10.2174/157015912803217341.
25. PMID: 21933950. Jankovic J. Diagnosis and treatment of psychogenic parkinsonism. J Neurol Neurosurg Psychiatry. 2011 Dec;82(12):1300-3. doi: 10.1136/jnnp-2011-300876. Epub 2011 Sep 20.
26. PMID: 31501181. Grimes D, et al. Canadian guideline for Parkinson’s disease. CMAJ. 2019 Sep 9;191(36):E989-E1004. doi: 10.1503/cmaj.181504.
27. PMID: 31427383. Simonet C, Tolosa E, Camara A, Valldeoriola F. Emergencies and critical issues in Parkinson’s disease. Pract Neurol. 2020 Feb;20(1):15-25. doi: 10.1136/practneurol-2018-002075. Epub 2019 Aug 19.
28. PMID: 15822109. Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations. Mov Disord. 2005;20 Suppl 11:S11-6. doi: 10.1002/mds.20458.
29. PMID: 17266092. Ravina B, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007 Jun 15;22(8):1061-8. doi: 10.1002/mds.21382.
30. PMID: 20717751. Ross JC, Cook AM, Stewart GL, Fahy BG. Acute intrathecal baclofen withdrawal: a brief review of treatment options. Neurocrit Care. 2011 Feb;14(1):103-8. doi: 10.1007/s12028-010-9422-6.
31. PMID: 34158937. Romito JW, Turner ER, Rosener JA, Coldiron L, Udipi A, Nohrn L, Tausiani J, Romito BT. Baclofen therapeutics, toxicity, and withdrawal: A narrative review. SAGE Open Med. 2021 Jun 3;9:20503121211022197. doi: 10.1177/20503121211022197.
32. PMID: 25432728. Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin. 2015 Feb;33(1):153-74. doi: 10.1016/j.ncl.2014.09.011.
33. PMID: 28461905. Termsarasab P, Frucht SJ. Dystonic storm: a practical clinical and video review. J Clin Mov Disord. 2017 Apr 28;4:10. doi: 10.1186/s40734-017-0057-z.
34. PMID: 26185277. Hawthorne JM, Caley CF. Extrapyramidal Reactions Associated with Serotonergic Antidepressants. Ann Pharmacother. 2015 Oct;49(10):1136-52. doi: 10.1177/1060028015594812. Epub 2015 Jul 16.
35. PMID: 25607477. Tekin U, Soyata AZ, Oflaz S. Acute focal dystonic reaction after acute methylphenidate treatment in an adolescent patient. J Clin Psychopharmacol. 2015 Apr;35(2):209-11. doi: 10.1097/JCP.0000000000000266.
36. PMID: 23649720. Albanese A, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013 Jun 15;28(7):863-73. doi: 10.1002/mds.25475. Epub 2013 May 6.
37. PMID: 31032123. Yaginuma K, et al. Haloperidol-Induced Dystonia due to Sedation for Upper Gastrointestinal Endoscopy: A Pediatric Case Report. Case Rep Emerg Med. 2019 Mar 27;2019:3591258. doi: 10.1155/2019/3591258.
38. PMID: 28461905. Termsarasab P, Frucht SJ. Dystonic storm: a practical clinical and video review. J Clin Mov Disord. 2017 Apr 28;4:10. doi: 10.1186/s40734-017-0057-z.
39. PMID: 24304390. Allen NM, Lin JP, Lynch T, King MD. Status dystonicus: a practice guide. Dev Med Child Neurol. 2014 Feb;56(2):105-12. doi: 10.1111/dmcn.12339. Epub 2013 Dec 4.
40. PMID: 20838215. Stryjer R, et al. Trazodone for the treatment of neuroleptic-induced acute akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010 Sep-Oct;33(5):219-22. doi: 10.1097/WNF.0b013e3181ee7f63.
41. PMID: 28168537. Cossu G, Colosimo C. Hyperkinetic Movement Disorder Emergencies. Curr Neurol Neurosci Rep. 2017 Jan;17(1):6. doi: 10.1007/s11910-017-0712-7.
42. PMID: 18024776. Wild EJ, Tabrizi SJ. The differential diagnosis of chorea. Pract Neurol. 2007 Nov;7(6):360-73. doi: 10.1136/pn.2007.134585.
43. PMID: 21670395. Robottom BJ, Factor SA, Weiner WJ. Movement disorders emergencies Part 2: hyperkinetic disorders. Arch Neurol. 2011 Jun;68(6):719-24. doi: 10.1001/archneurol.2011.117.
44. PMID: 34446993. Chandarana M, Saraf U, Divya KP, Krishnan S, Kishore A. Myoclonus- A Review. Ann Indian Acad Neurol. 2021 May-Jun;24(3):327-338. doi: 10.4103/aian.AIAN_1180_20. Epub 2021 May 21.
45. PMID: 31356293. Caviness JN. Myoclonus. Continuum (Minneap Minn). 2019 Aug;25(4):1055-1080. doi: 10.1212/CON.0000000000000750.
46. PMID: 25438253. Sandroni C, et al. Prognostication in comatose survivors of cardiac arrest: an advisory statement from the European Resuscitation Council and the European Society of Intensive Care Medicine. Resuscitation. 2014 Dec;85(12):1779-89. doi: 10.1016/j.resuscitation.2014.08.011.
47. PMID: 33765189. Nolan JP, et al. European Resuscitation Council and European Society of Intensive Care Medicine guidelines 2021: post-resuscitation care. Intensive Care Med. 2021 Apr;47(4):369-421. doi: 10.1007/s00134-021-06368-4. Epub 2021 Mar 25.
48. PMID: 25511790. Baizabal-Carvallo JF, Jankovic J. Stiff-person syndrome: insights into a complex autoimmune disorder. J Neurol Neurosurg Psychiatry. 2015 Aug;86(8):840-8. doi: 10.1136/jnnp-2014-309201. Epub 2014 Dec 15.
49. PMID: 17385093. Andreadou E, Kattoulas E, Sfagos C, Vassilopoulos D. Stiff person syndrome: avoiding misdiagnosis. Neurol Sci. 2007 Mar;28(1):35-7. doi: 10.1007/s10072-007-0745-9.
50. PMID: 21357910. Afshar M, Raju M, Ansell D, Bleck TP. Narrative review: tetanus-a health threat after natural disasters in developing countries. Ann Intern Med. 2011 Mar 1;154(5):329-35. doi 10.7326/0003-4819-154-5-201103010-00007.
51. PMID: 3652717. Lipman J, James MF, Erskine J, Plit ML, Eidelman J, Esser JD. Autonomic dysfunction in severe tetanus: magnesium sulfate as an adjunct to deep sedation. Crit Care Med. 1987 Oct;15(10):987-8.
52. PMID: 17055945. Thwaites CL, Yen LM, Loan HT, Thuy TT, Thwaites GE, Stepniewska K, Soni N, White NJ, Farrar JJ. Magnesium sulfate for treatment of severe tetanus: a randomized controlled trial. Lancet. 2006 Oct 21;368(9545):1436-43. doi: 10.1016/S0140-6736(06)69444-0.

Add comment