


1 November, 2022 by Shahriar Lahouti.

CONTENTS
- Preface
- Neuroanatomy of motor pathway
- Initial approach
- Evaluation
- Management
- Specific conditions
- Appendix
- Media
- Going further
- References
Preface
Weakness is a common ED presentation, with up to 10% of ED patients presenting with this chief complaint. Because of the vague nature of this presentation (i.e. weakness), there is an increased risk of misdiagnosis, hospital admission, and death.
- In a study of adult patients presenting to an ED for weakness, the majority had generalized weakness (65%) as opposed to focal weakness (35%) *. The rate of incorrect ED diagnoses in patients with vague symptoms such as generalized weakness was found to be as high as 50% in certain studies *.
The approach to the diagnosis and initial management of patients presenting to the ED with acute, nontraumatic neurologic and neuromuscular weakness will be reviewed in this post.
Neuroanatomy of motor pathways
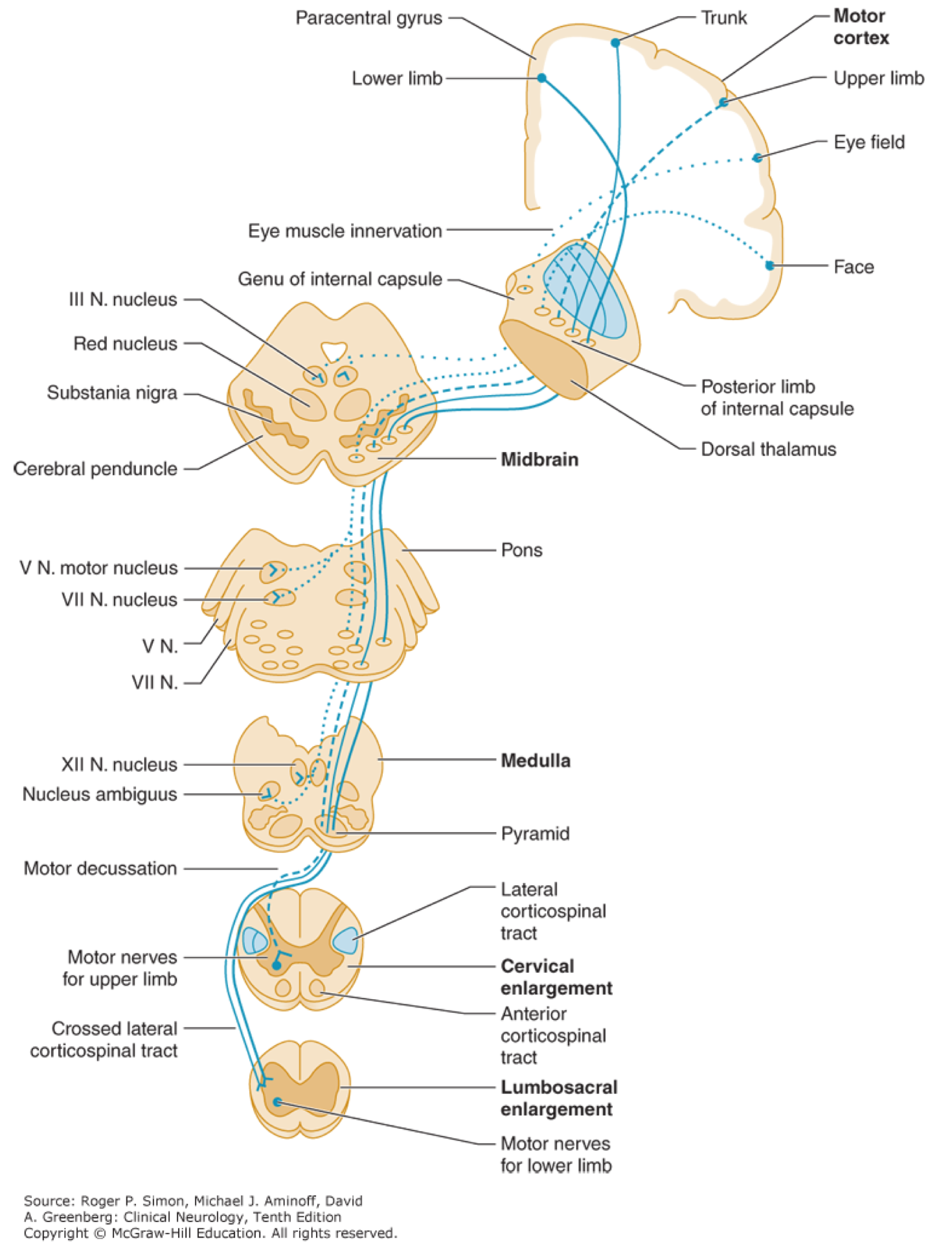
Corticospinal tract (pyramidal tract)
- Upper motor neuron cell bodies lie in the motor cortex (within the precentral gyrus).
- Their axons travel in the subcortical white matter within the posterior limb of the internal capsule.
- They run through the cerebral peduncles into the ventral/anterior brainstem.
- They cross (decussate) at the junction of the medulla and the cervical spinal cord.
- They run down the posterolateral spinal cord.
- Finally, upper motor neuron axons synapse onto lower motor nerves within the anterior horn of the spinal cord grey matter.
Corticobulbar tract
- Primarily involved in carrying the motor function of the non-oculomotor cranial nerves (e.g. cranial nerves V, VII, IX, XII, and motor region of cranial nerve X.
- The corticobulbar tract originates in the primary motor cortex of frontal lobe, just superior to the lateral fissure and rostral to the central sulcus in the precentral gyrus.
- It descends via corona radiata and genue of internal capsule with a few fibers in the posterior limb of internal capsule.
- In the midbrain the internal capsule becomes the cerebral peduncle.
- The corticobulbar fibers exit at the appropriate level of the brainstem to synapse on the lower motor neurons of the cranial nerves ( V, VII, IX, XII, the motor regions of cranial nerve X in the nucleus ambiguus.
- The corticobulbar tract innervates cranial motor nuclei bilaterally with the exception of the lower facial nuclei (which innervates facial muscles below the eyes) and the genioglossus muscle, which are innervated only unilaterally by the contralateral cortex.
Initial approach
History
- Distinguish true weakness (neuromuscular) from subjective fatigue.
- Assess the change in the patient’s functional status compared with their baseline
- What activities/movements do you have trouble with?
- Assess for other associated symptoms, exacerbating or mitigating factors, and recent lifestyle or medication changes.
- Preceding prodromal illness?
- Exposures? (e.g. mosquitoes, unusual food, travel, intravenous drug use)
- Fever?
- Comorbidities?
-
Assess for psychiatric conditions such as depression.
Localization of pathology and identifying the cause
- Distribution of weakness
- Bilateral vs. unilateral weakness?
- Upper vs. lower extremities?
- Myotomal pattern of weakness distribution?
- Is the description of weakness consistent with a particular peripheral nerve?
- Ocular (e.g. double vision, droopy eyelids) and bulbar involvement (e.g. voice change)?
- Shortness of breath?
- Duration?
- Acute (seconds to minutes): CVA, ICH, hypoglycemia, poisoning, spinal cord syndromes, periodic paralysis.
- Subacute (hours to days): Botulism, tetanus, Myasthenia gravis crisis, GBS, transverse myelitis, electrolytes (hypokalemia, hyponatremia), conversion disorder
- Chronic (weeks to months): Amyotrophic lateral sclerosis, psychiatric disorders (e.g. depression)
- Pattern:
- Proximal vs. distal weakness?
- Does the degree of weakness fluctuate?
- Associated signs and symptoms
- Altered level of consciousness?
- Cortical findings (e.g. aphasia, hemineglect, agnosia or apraxia, gaze preference, visual deficit)?
- Sensory abnormality (e.g. pain, numbness, paresthesia)?
- Muscle cramping or aching?
- Bowel and/or bladder symptoms?
Physical examination
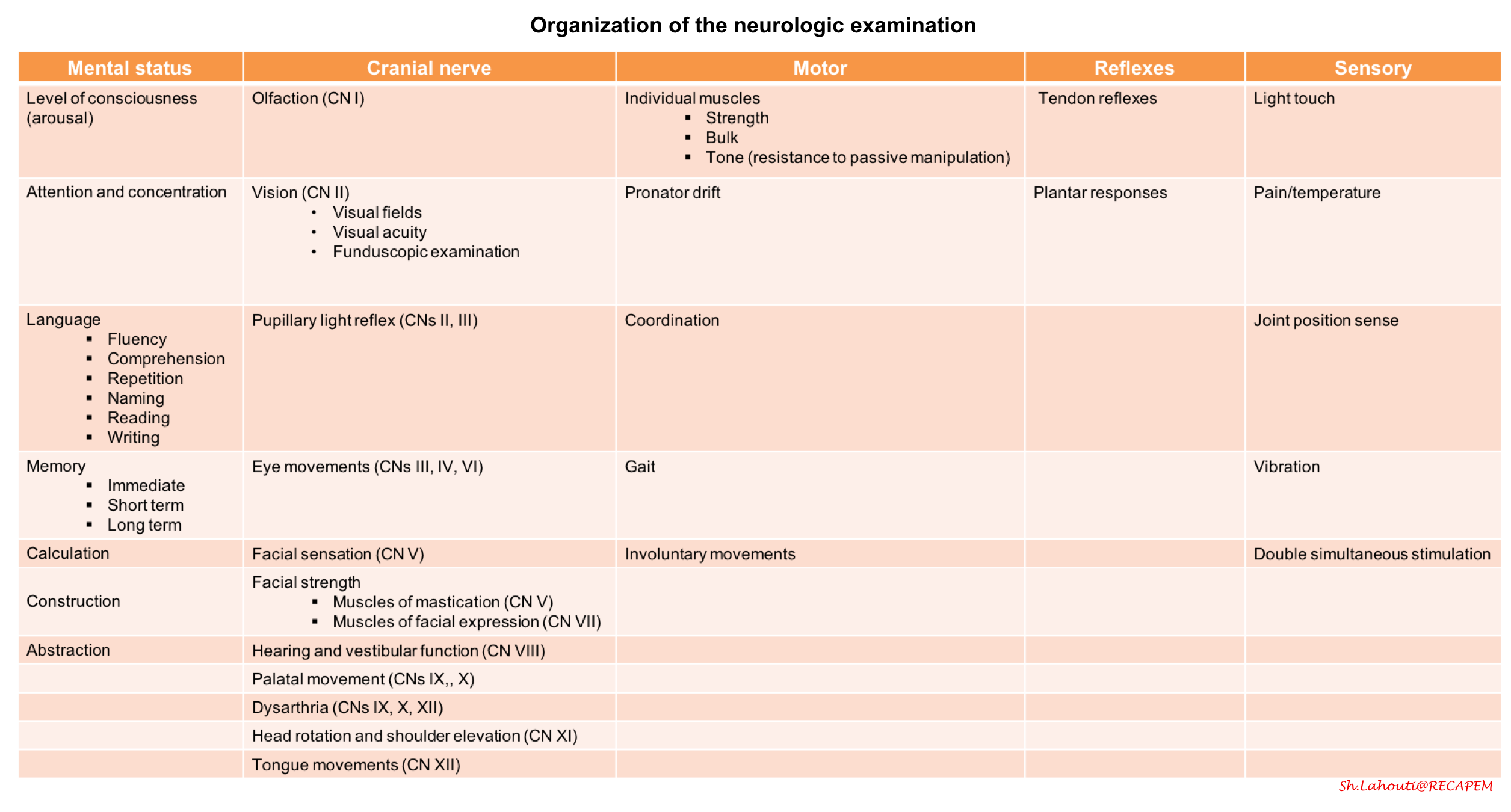
- Complete set of vitals, including temperature and glucose
-
Screening neurological exam (here)
-
Mental status
- Level of consciousness (arousal)
- Level of orientation to person, place, and time.
- Make sure they can follow at least one complicated command. If their responses are appropriate and they are able to relate a detailed and coherent medical history, no further mental status testing is necessary (unless they have cognitive complaints).
-
Cranial nerve abnormalities
-
Test visual fields in one eye, both pupillary responses to light, eye movements in all directions, facial strength, and hearing to finger rub.
-
- Sensory examination
- Test light touch sensation in all four distal limbs, including double simultaneous stimulation. Test vibration sense at the great toes.
- Motor system
- Assess muscle strength, tone and bulk.
- Pronator drift (here)
- Reflexes: The Neuroanatomy of commonly tested reflexes:
- Biceps: C5-C6.
- Brachioradialis: C6.
- Triceps: C7.
- Patellar: L4.
- Achilles: S1.
- Clonus (here)
- Babinski sign (plantar responses); here.
- Hoffmann sign (here)
- Coordination
- Gait
- Rapid alternating movements
- Finger-to-nose testing, and heel-to-shin testing.
-
Localizing weakness
One useful systematic approach to the various lesions of the neuromuscular system that can cause true muscle weakness is to place them into categories determined by the organization of the neuromuscular system. This include motor cortex, corticospinal tracts, anterior horn cells, spinal nerve roots, peripheral nerves, neuromuscular junction, and finally muscle.
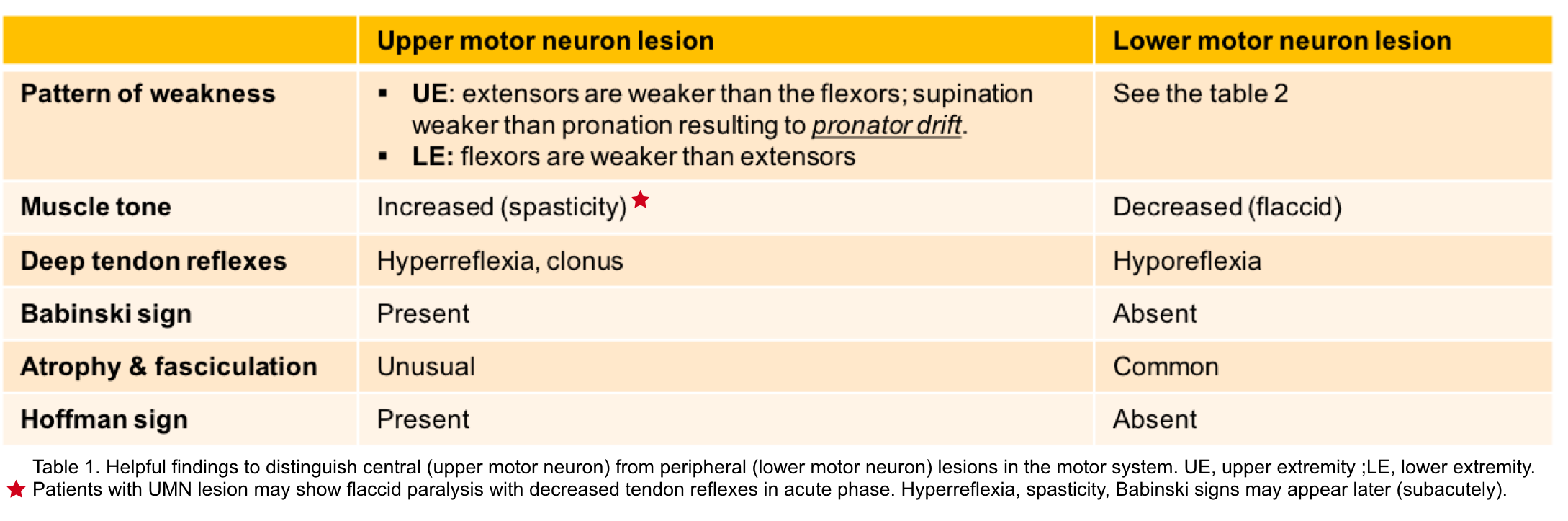
Upper motor neurons
- They are found in the cerebral cortex and brainstem and carry information down to activate interneurons and lower motor neurons.
- UMN lesions occurs in the neural pathway above the anterior horn of spinal cord. The resulting changes in neuromuscular performance are presented in table 1 below *.
Lower motor neurons
- LMN are located in either the anterior nerve roots (spinal lower motor neurons) or the cranial nerve nuclei of brainstem and cranial nerves with motor function (cranial nerve lower motor neurons). Cranial nerve lower motor neurons control movements of the eyes, face and tongue, and contribute to chewing, swallowing and vocalization.
- The characteristic neuromuscular findings with LMN lesion are listed in table 1 below.
Motor unit
- A motor unit is made up of a motor neuron and all of the skeletal muscle fibers innervated by the neuron’s axon terminals, including the neuromuscular junctions (NMJ) between the neuron and the fibers.
- The characteristic features helpful to differentiate peripheral neuropathy from NMJ disorders vs, myopathy is shown in table 2 below.

- In patient with weakness, an algorithmic approach based upon a thorough history and examination facilitates neuroanatomic localization of the pathology. The steps towards localizing the weakness is discussed in detail below.
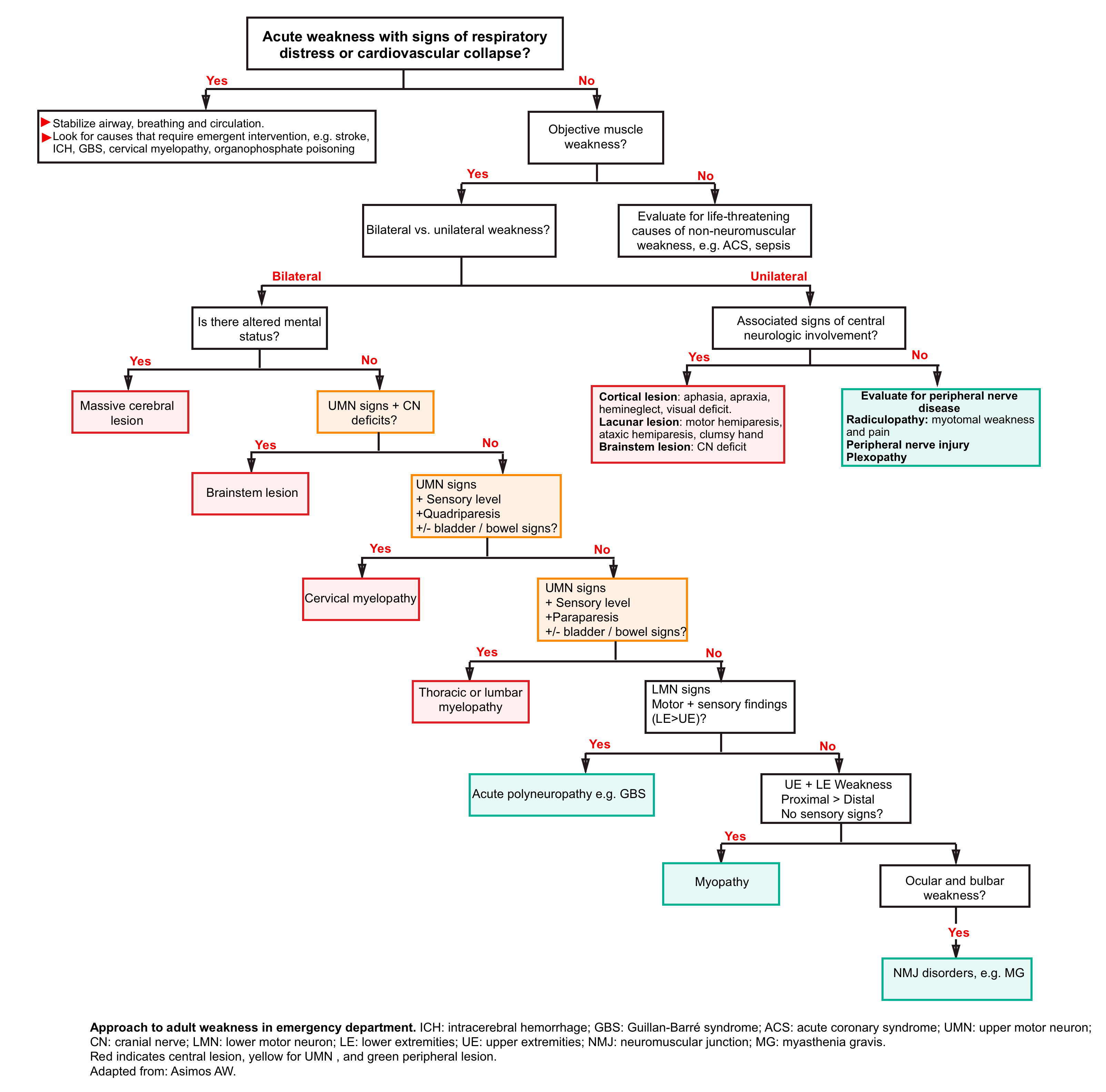
Principles of localizing the lesion
#1: Determine the pattern of muscle weakness:
- Generalized weakness
- Neurologic causes:
- Myasthenia gravis
- Periodic paralysis
- Advanced disuse atrophy from prolonged bed rest
- Muscle wasting from malignancy
- Longstanding motor neuron disease.
- Critical non-neurological causes
- Sepsis
- Acute cardiovascular disease (eg, coronary syndrome, decompensated heart failure)
- Poisoning e.g. organophosphate and carbamate poisoning, botulism, and carbon monoxide (CO) poisoning.
- Severe electrolyte abnormalities (eg, hypokalemia, hyperkalemia, hyponatremia)
- Endocrine crisis e.g. diabetic ketoacidosis, hypoadrenal (Addisonian) crisis, myxedema coma, thyrotoxicosis.
- Neurologic causes:
- Non-generalized weakness: it can be characterized as unilateral or bilateral (figure below).
- Unilateral weakness most likely reflects disease of the central or peripheral nervous system (rather than NMJ or myopathic disorders).
- Are cortical signs (e.g. aphasia, neglect, agnosia, or apraxia) present?
- If so, pathology lies in the cerebral cortex.
- Is the face involved (eg, facial droop)?
- Generally, unilateral facial weakness implies a lesion above the spinal cord, either in the brainstem or cortex.
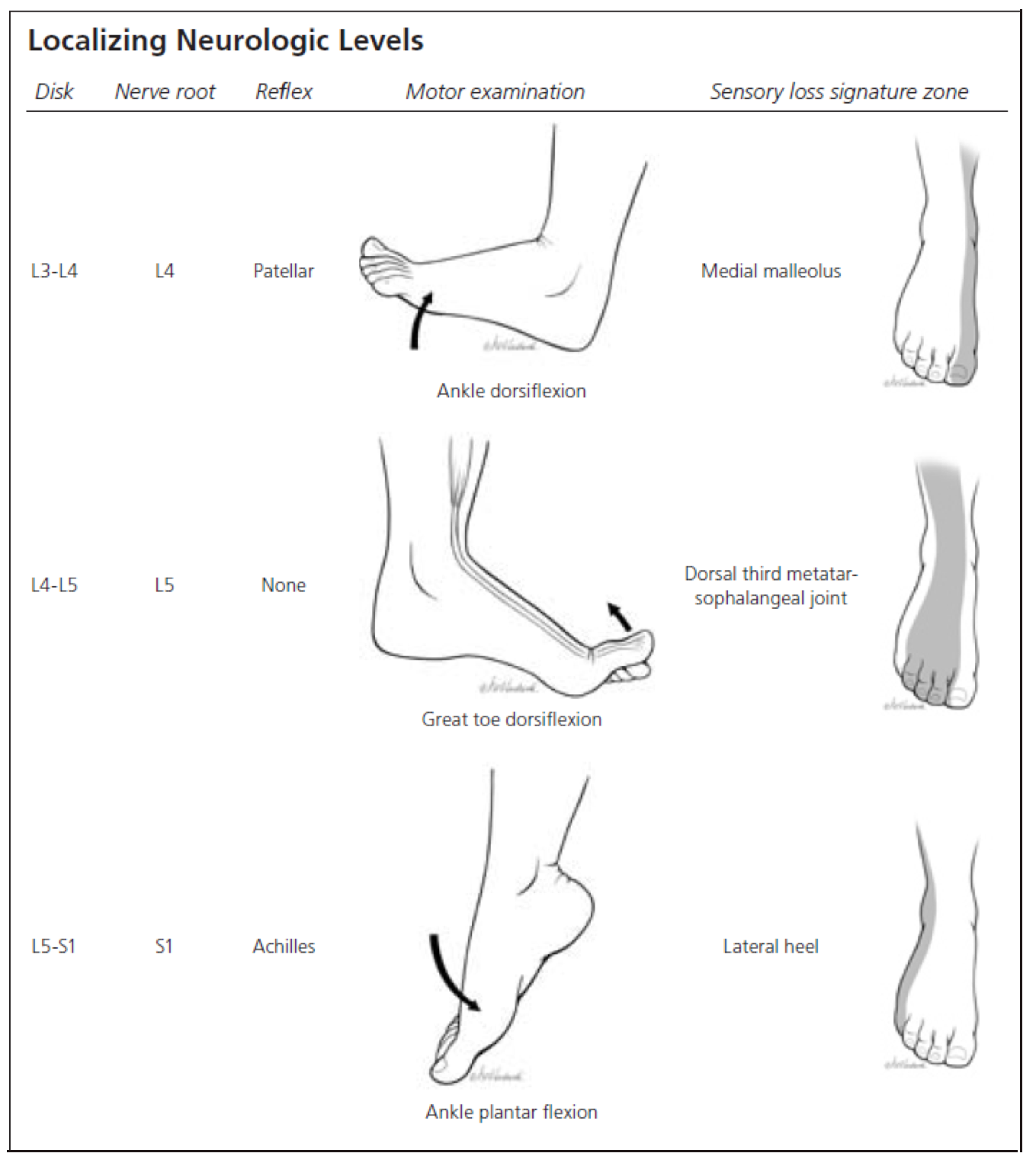
- Is there a myotomal pattern to the distribution of weakness? (appendix 1)
- Is the description of weakness consistent with a particular peripheral nerve? (appendix 2)
- Are cortical signs (e.g. aphasia, neglect, agnosia, or apraxia) present?
- Bilateral weakness
- Is mental status depressed?
- A diminished mental status in the presence of bilateral weakness suggests a central nervous system lesion.
- Are there cranial nerves deficit (involving tongue, jaw, face, or larynx)?
- Presence of CN deficit and bilateral weakness is suggestive for brainstem lesions.
- Is there sensory involvement? If so, is a sensory level deficit suggested?
- A sensory deficit demarcated by a specific dermatome suggests spinal cord pathology. (appendix 3)
- A more diffuse sensory findings such as paresthesia may be an early sign of a polyneuropathy.
- Is there bladder involvement?
- Bladder dysfunction suggests a myelopathy.
- Does weakness primarily involve proximal or distal muscles? See table 2 above.
- Involvement primarily of proximal muscles suggests a myopathy or NMJ disorders, while involvement of distal muscles suggests a polyneuropathy (or early motor neuron disease).
- Is there sensory involvement?
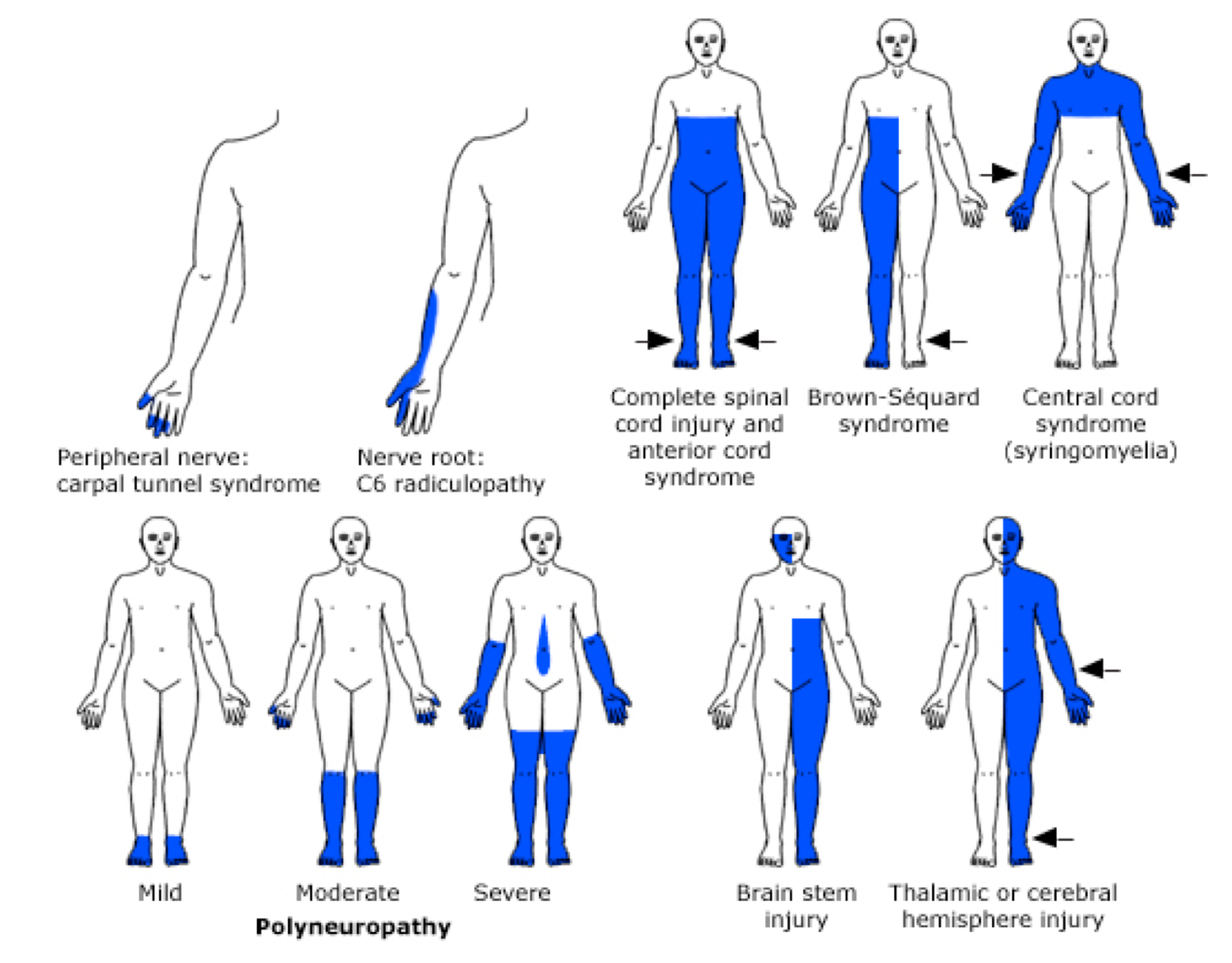
- Distribution and pattern of sensory deficit can suggest site of the lesion (figure below). More on sensory deficit here.
- Is there eye muscle or “bulbar” weakness? This may happen in
- Cerebral cortical lesions
- Brain stem lesions.
- Myasthenia gravis.
- Botulism.
- Tick paralysis.
- Miller Fisher variant of GBS.
- Does the degree of weakness fluctuate?
- Worsening of weakness with repeated activity such as chewing or maintaining upward gaze suggests NMJ disorder e.g. MG.
- Is mental status depressed?
- Unilateral weakness most likely reflects disease of the central or peripheral nervous system (rather than NMJ or myopathic disorders).
#2. Determining the cause of the lesion.
- Once the neuromuscular site of the lesion causing weakness has been identified, the disorders at each site can be categorized more specifically based on constellation of signs and symptoms. This is discussed in the following section (differential diagnosis).
Distribution and pattern of weakness
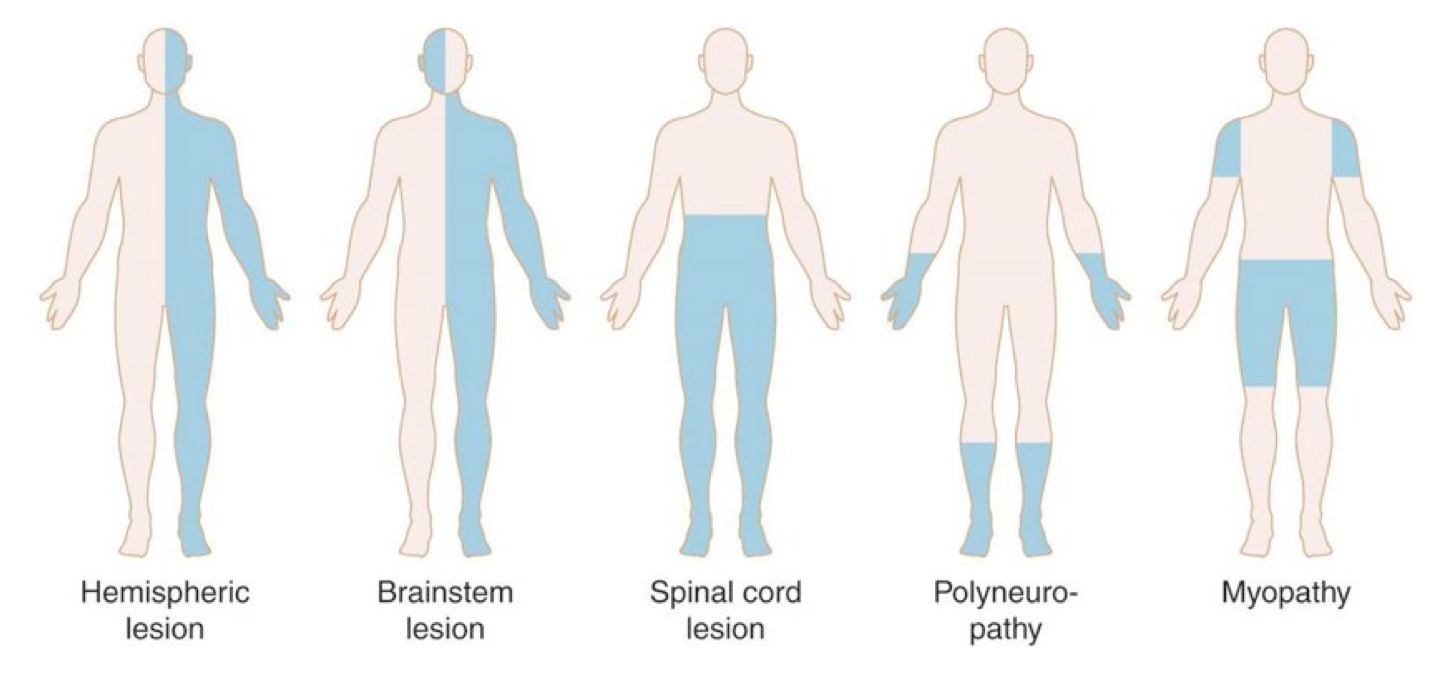
Distribution and pattern of sensory deficits
Differential diagnosis
👉Central
- Cerebrum
- Typical findings:
- Cortical deficit (+/- altered mental status).
- Often distal > proximal weakness.
- Often unilateral weakness.
- Upper motor neuron signs may appear, especially subacutely (e.g. hyperreflexia, spasticity, Babinski sign).
- Cranial nerve deficit may occur (although cranial nerve abnormalities can also occur with other etiologies).
- DDx
- Typical findings:
- Subcortical / brainstem
- Typical findings
- Typically cortical deficits are absent.
- Weakness syndromes referable to the brainstem may present with “crossed” findings (ipsilateral cranial nerve weakness and contralateral hemiparesis) due to lesions involving cranial nerve nuclei or their tracts and the corticospinal tract before its decussation.
- However, this is often not the case. As an example, a lacunar stroke in the pons can appear indistinguishable clinically (hemiparesis, ataxia) from a lacunar stroke affecting the internal capsule.
- Brainstem lesions can also invariably cause typical neurologic signs and symptoms such as vertigo, depressed mental status, visual field deficits, oculomotor abnormalities, and bulbar findings.
- DDx
- Lacunar infarction
- Structural lesion
- Multiple sclerosis
- Typical findings
- Spinal cord lesions (myelopathies) with involvement of multiple tract
- Typical findings
- Neurologic abnormalities may localize to a spinal level.
- No cranial nerve abnormalities (e.g. No bulbar or cervical neck weakness).
- Sensation is frequently involved.
- Bowel and bladder dysfunction (urinary retention or incontinence depending on the level of spinal cord injury) may occur.
- Disturbances in autonomic function, such as loss of sweating, trophic skin changes, loss of temperature control, and vasomotor instability, can occur below the level of the lesion
- Upper motor neuron signs may appear, especially subacutely (e.g. hyperreflexia, spasticity, Babinski sign). Acutely, patients may have transient spinal shock, with loss of spinal function below the level of the lesion and areflexia.
- DDx
- Acute transverse myelitis
- Spinal cord infarct
- Spinal cord compression (e.g. trauma, epidural abscess, malignancy, central intervertebral disc herniation) *.
- Multiple sclerosis
- Typical findings
- Spinal cord: selective involvement of anterior horn cells (acute flaccid myelitis)
-
- Typical clinical findings:
- Variable patterns of weakness (often asymmetric; may have both proximal and distal weakness).
- Reflexes are usually reduced.
- Sensation is unaffected.
- DDx:
- Poliomyelitis
- Paraneoplastic syndrome
- Amyotrophic lateral sclerosis
- Spinal muscular atrophy
- Typical clinical findings:
👉Peripheral
- Peripheral polyneuropathy and/or polyradiculopathy
- Typical clinical findings:
- Ascending weakness (distal > proximal).
- Bulbar involvement may occur, but ocular involvement is rare.
- Sensation is frequently involved.
- Reflexes are reduced.
- Lower motor neuron findings may occur (atrophy, fasciculations).
- DDx *
- Guillain-Barre syndrome (more on this below).
- Diabetic neuropathy
- Lyme disease.
- Vitamin deficiency (e.g. thiamine deficiency; B12 deficiency or nitrous oxide poisoning).
- Vasculitic neuropathy (e.g. rheumatoid arthritis, polyarteritis nodosa).
- Acute intermittent porphyria.
- Toxins: Heavy metals (e.g. arsenic, mercury), ethylene glycol, methanol.
- Critical illness polyneuropathy.
- Leptomeningeal malignancy.
- Typical clinical findings:
- Peripheral neuropathy (mononeuropathy)
- Typical findings
- Loss of motor and sensory function within the distribution of the corresponding nerve.
- Reproduction of symptoms with movement (compressive neuropathy)
- DDx
- Nerve compression syndrome
- Bell’s palsy (see below)
- DM
- Typical findings
- Radiculopathy
- Typical findings
- Pain or numbness in a dermatomal pattern accompanied by weakness in a segmental pattern suggests a radiculopathy.
- Unilateral absence of a specific lower extremity reflex. As an example, unilateral loss of the ankle jerk suggests a lesion at the S1 nerve root.
- DDx
- Degenerative disc disease
- Spinal epidural abscess / hematoma
- Infections such as shingles
- Typical findings
- Neuromuscular junction
- Typical clinical findings:
-
- Fatigability, fluctuation in weakness.
- May see proximal limb and neck weakness (similar to myopathy), or descending weakness.
- Frequent ocular (e.g. ptosis, diplopia, ophthalmoplegia) and bulbar involvement (e.g. dysarthria, dysphagia).
- Symmetric weakness.
- Sensation is intact.
- Reflexes are normal (unless weakness is severe).
-
- DDx *
- Myasthenia gravis (more on this below).
- Medications: neuromuscular blocking agents, aminoglycosides, anticholinesterase inhibitors
- Botulism (here).
- Tetanus.
- Scorpion sting, Shellfish poisoning, Snake venom.
- Tick paralysis (toxin interferes with acetylcholine release).
- Organophosphate poisoning.
- Hypermagnesemia.
- Typical clinical findings:
- Myopathy
- Typical findings
- Proximal limb and neck weakness.
- Symmetric weakness.
- Reflexes are preserved (unless weakness is severe).
- Atrophy may occur (but without fasciculations, as might be seen in lower motor neuron disease).
- Sensation is intact, but patients may have cramps/myalgias/stiffness.
- Muscle tenderness may occur.
- DDx
- Inflammatory myositis (e.g. polymyositis, dermatomyositis, lupus, scleroderma).
- Electrolyte induced: Hypo/hyperkalemia, hypomagnesemia, hypophosphatemia.
- Endocrine related: Hypo/hyperthyroidism.
- Drug-induced toxic myopathy (e.g. alcohol, chloroquine, cocaine, colchicine, cyclosporine, statins, steroid).
- Critical illness myopathy.
- Mitochondrial disease.
- Muscular dystrophy
- Muscle ion channelopathies: Periodic paralysis
- Rhabdomyolysis
- Typical findings
👉Non-neuromuscular
- Physiologic
- Hypoperfusion: Cardiac ischemia, heart failure, hypovolemia, anemia
- Metabolic: Hypoglycemia, hyponatremia, hypokalemia, diabetic ketoacidosis, myxedema coma, thyrotoxicosis, adrenal insufficiency
-
Poisoning: sedative-hypnotic toxicity, opioid overdose, stimulants withdrawal, CO poisoning, organophosphate, digoxin overdose, heavy metals
- Infection: Sepsis, occult infection, chronic infection
- Malignancy
- Arthritis
- Non-physiologic / non-categorical
- Typical findings:
- Weakness is variable, inconsistent, and with a pattern of distribution that cannot be explained on a neuroanatomic basis.
- The severity of weakness is out of keeping with the patient’s daily activities.
- Babinski sign is absent.
- On formal testing, antagonists may contract when the patient is supposedly activating the agonist muscle.
- DDx
- Chronic fatigue syndrome, depression, anxiety disorder.
- Conversion disorder
- Patient with conversion disorder can present with nonepileptic seizures, weakness and paralysis, sensory complaints, speech disturbances, globus sensation, visual /cognitive symptoms, and movement disorders.
- Mixed symptoms could be present in 35% of patients.
- Onset of conversion symptoms is often sudden (≤6 hours to maximal onset).
- Certain clinical features are commonly observed at onset of the conversion symptoms including panic attacks, migraine headache, or pain, as well as physical injury.
- Helpful tips on examination
- Symptoms do not conform to known anatomical pathways and physiologic mechanisms (incongruity between the symptoms and recognized disease).
- Inconsistency at different points in the examination (e.g. a patient with no ankle plantar flexion while supine on the examination table is able to stand on tip toes).
- Patients with sensory symptoms describes loss of sensation in midline (midline splitting) *. True neurogenic sensory loss does not follow midline splitting in which sensation is split exactly in the midline; however, midline splitting can occur in thalamic stroke.
- Patients with weakness and paralysis frequently have positive Hoover’s sign on examination *.
- Typical findings:
Evaluation
- Blood gas measurement
- In patients with neuromuscular disease, arterial blood gas measurement may not be helpful in establishing the likelihood of imminent respiratory failure, because pCO2 can rise quickly as diaphragmatic and intercostal muscle weakness progresses *.
- Such patients commonly lose tidal volume before upper airway weakness develops, resulting in an increased respiratory rate to maintain minute ventilation. In such cases, pCO2 may remain normal or even low until the tidal volume becomes dangerously low. Therefore blood gas measurement has little role in most patients with acute neuromuscular weakness, and selected pulmonary function tests are needed to determine the degree of respiratory compromise.
- Patients without chronic respiratory dysfunction should have a normal respiratory drive (unless they are on medications that block the respiratory drive e.g. opioids). These patients shouldn’t become hypercapnic until they are totally exhausted and frankly dying.
- Patients with chronic hypercapnia (e.g. due to severe COPD) don’t have a normal respiratory drive. These patients may develop insidiously worsening hypercapnia without looking distressed.
- Rather than developing respiratory extremis, these patients may quietly accumulate CO2 and become sleepy (due to CO2 narcosis). Therefore blood gas measurement is useful in patients with altered mental status plus chronic hypercapnia.
- In patients with neuromuscular disease, arterial blood gas measurement may not be helpful in establishing the likelihood of imminent respiratory failure, because pCO2 can rise quickly as diaphragmatic and intercostal muscle weakness progresses *.
- Bedside pulmonary function tests
- Methods:
- Forced vital capacity (FVC)
- It is the largest volume breath the patient is able to take.
- FVC is an integrated reflection of multiple parameters: inspiratory strength, expiratory strength, and lung compliance. Therefore it is a better predictor of respiratory failure than the negative inspiratory force (a.k.a. maximal inspiratory pressure, which measures only diaphragmatic strength).
- Single breath count test
- May be used as a surrogate for the forced vital capacity (FVC) *.
- The patient should take the deepest possible breath and then count upwards from one as high as they can.
- Patients must count at a rate of about two per second (120 per minute).
- A count >25 may suggest a reasonably preserved forced vital capacity (over ~2 liters).
- Forced vital capacity (FVC)
- Indications:
- Initial diagnosis of neuromuscular weakness. In a patient with respiratory failure of unclear etiology, pulmonary function tests may be helpful by identifying the presence of neuromuscular weakness.
- Initial evaluation and triage of patients with known neuromuscular weakness.
- If admission FVC is normal and the patient is dyspneic, this suggests that something else is going on (e.g. pulmonary embolism).
- If the admission FVC is significantly low (e.g. < ~30 cc/kg), this supports the need for a higher intensity of respiratory monitoring.
- Monitoring the patient
- Intermittently measuring the FVC may help determine how the patient is responding to therapy.
- Measuring the FVC about three times per day is probably adequate.
- To be significant (e.g. decrease of >30% of vital capacity), changes should represent a consistent trend over several measurements (not just one aberrant measurement).
- Methods:
- Blood tests:
- CBC
-
Point-of-care blood glucose
- Electrolytes (including Ca/Mg/Phos).
- Creatine kinase elevation may suggest myopathy.
- Consider screening for HIV, if this is a possibility.
- TSH (thyroid-stimulating hormone) may be considered.
- Other labs may be requested when clinical diagnosis is suspected such as
- ESR (polymyalgia rheumatica and other autoimmune processes)
-
Carboxyhemoglobin (carbon monoxide poisoning)
-
Digoxin level (cardiac glycoside toxicity)
-
Ammonia level (hepatic encephalopathy)
-
Toxicology screen
- Infection (urinalysis / cultures, CXR
- ECG may be useful in the presence of certain electrolyte disturbances, e.g. hypokalemia.
- Lumbar puncture
- CSF is generally normal in myopathy, neuromuscular junction disorders and peripheral neuropathies (although CSF abnormalities may occur in neuropathies which involve the nerve roots such as Guillain-Barre syndrome).
- CSF analysis may be helpful in following conditions:
- Guillain-Barré syndrome: classically causes albuminocytologic dissociation (elevated protein, despite a normal cell count<5/uL, and erythrocyte count <50/uL) *.
- Myelitis. CSF analysis is normal in approximately half of patients. In the other half, it may reveal an elevated protein or moderate lymphocytosis, but the glucose concentration remains normal.
- Neuroradiology
-
Head computed tomography (CT)
-
Consider neuroimaging in all cases of focal weakness.
-
Consider a non-contrast study if stroke, intracranial masses, intracranial bleed, or cerebral edema are suspected.
-
Consider a contrast study if tumor, metastatic lesions, or specific infection (such as cerebral abscess) are suspected.
-
Consider magnetic resonance imaging (MRI) if a mass is suggested on CT.
-
-
-
CT angiography head/neck
-
Consider if aneurysm, arteriovenous malformation, venous sinus thrombosis, or vertebrobasilar insufficiency are suspected.
-
-
MRI
-
Consider if cerebrovascular accident, spinal cord infarct, cauda equina, transverse myelitis are suspected
-
-
- EMG and nerve conduction study
Approach to undifferentiated weakness in critical patients
Algorithmic approach to the adult patients with weakness in the emergency department is represented above.
-
Place the patient on a cardiac monitor and continuous pulse oximetry.
-
Secure intravenous access.
-
Obtain point-of-care glucose.
STEP 1. Assess ABC.
- The immediate life threats from acute neuromuscular weakness include inability to protect or maintain the airway, respiratory failure from thoracic and diaphragmatic muscle weakness, and circulatory collapse from autonomic instability.
-
Airway
-
Consider reversible conditions (hypoglycemia and opioid toxicity) before intubation.
-
Factors to consider in the decision to intubate *
-
Increasing muscle weakness
-
Dysphagia or dysphonia (weak or muffled voice)
-
Dyspnea on exertion and at rest or orthopnea
-
Rapid, shallow breathing: In patients with neuromuscular disease, tachypnea often presents sooner than, and may herald other signs of impending respiratory failure.
-
Weak cough
-
Interrupted speech (gasping for air)
-
Use of accessory muscles
-
Paradoxical (abdominal) breathing
-
Weakness of trapezius/neck muscles (neck flexors): inability to lift head from bed
-
Cough after swallowing
-
Decreased level of consciousness
-
Have a lower threshold to control the airway if the patient requires transfer or movement to unmonitored areas.
-
-
Hypoxemia or hypercapnia (occurs late)
-
Forced vital capacity <15 mL/kg, or declining one breath count
-
-
A non-depolarizing paralytic (e.g. rocuronium) is recommended for rapid sequence intubation in undifferentiated patients, or if hyperkalemia is likely (e.g. renal failure, heat stroke, chronic neuromuscular disease).
-
- Breathing
- Provide oxygen by nasal cannula, non-rebreather mask, or non-invasive positive pressure ventilation as necessary.
- Maintain oxygen saturation >90%.
-
Circulation
-
Give intravenous fluids if there is evidence of hypotension/hypoperfusion.
-
-
Disability
-
Check for focal neurologic findings, including Glasgow Coma Scale, pupillary exam and deep tendon reflexes.
-
If glucose <55 mg/dL; give dextrose 50% in water 50 mL (25g) IV × 1, or dextrose 10% in water 250 mL (25 g) IV as rapid infusion.
-
STEP 2. Consider life-threatening causes of weakness in critically ill patients *
- Neurologic weakness
- Ischemic or hemorrhagic stroke, Subarachnoid hemorrhage (SAH)
- CNS infection
- Spinal cord injury (including compression) or inflammation
- Guillain-Barré syndrome
- Myasthenic crisis
- Important causes of generalized weakness to consider in the patient with respiratory distress or hemodynamic instability include:
- Sepsis
- Acute cardiovascular disease (eg, coronary syndrome, decompensated heart failure)
- Intoxication or poisoning e.g. organophosphate and carbamate poisoning, botulism, and carbon monoxide poisoning.
- Severe electrolyte abnormalities (eg, hypokalemia, hyperkalemia)
- Muscle weakness usually does not occur at potassium concentrations above 2.5 meq/L if hypokalemia develops slowly, but significant weakness may occur with sudden decreases. Weakness usually begins with the lower extremities, progresses to the trunk and upper extremities, and can worsen to the point of paralysis.
- Patients with renal dysfunction can develop hyperkalemia that manifests initially as weakness and may deteriorate into a life-threatening arrhythmia if untreated.
- Endocrine crisis (eg, DKA, hypoadrenal (Addisonian) crisis, myxedema coma, thyrotoxicosis)
STEP 3. Neuroanatomical approach to determine site and cause of the lesion (explained above).
STEP 4. Treatment is determined primarily by the underlying cause of the weakness. Some specific conditions are discussed below.
Guillain-Barré syndrome
- Acute polyneuropathy characterized by immune-mediated peripheral nerve myelin sheath or axon destruction.
- A trigger of GBS often occurs 5 days to 4 weeks before the onset of neurologic symptoms.
- Symptoms are at their worst in 2 to 4 weeks, and recovery can vary from weeks to a year.
- There are many variants of GBS. The demyelinating form involves lymphocytic infiltration of the myelin sheath of peripheral nerves and the axonal form involves motor paralysis with sensory function intact.
- The Miller-Fisher syndrome variant is characterized by ophthalmoplegia, cerebellar ataxia, and areflexia *. Extremity weakness can occur. However, patients usually don’t develop critical illness or respiratory failure.
- Clinical presentation and findings
- Preceding infection, followed by progressive (over ~2 weeks) ascending symmetric flaccid paralysis (may involve diaphragm) with loss of deep tendon reflexes. *
- Cranial nerve involvement (fascial weakness, dysphagia, dysarthria, ophthalmoplegia) is common.
- Sensory disturbance (paresthesia/pain) is often the first symptom. Sensory loss can occur, but this is mild compared to motor dysfunction.
- Findings localizing to the brain (except for Miller Fisher syndrome) should be absent.
- Autonomic dysfunction (tachycardia, bradycardia, dysrhythmias, wide variations in blood pressure, postural hypotension, urinary retention, constipation, facial flushing, anhydrosis, hypersalivation) may be present.
- Absence of fever at onset.
- Cytoalbuminologic dissociation of cerebrospinal fluid (high protein and low white cell count) *.
- 🚩Findings inconsistent with GBS *
-
- Maximum weakness reached within <24 hours or >4 weeks.
- Substantial asymmetry of weakness
- Severe sensory signs, with little or no weakness at onset.
- Absence of sensory symptoms (think anterior horn cell disease, neuromuscular, low potassium).
- Fever at onset.
- CSF white cell count >50/mm3.
- Neutrophils elevated.
- Sensory level suggestive of a focal spinal cord lesion.
- Bowel and bladder dysfunction at onset; severe and persistent bowel and bladder dysfunction (this may suggest myelopathy).
- Neurological deficit localized to the brain (except for Miller Fisher syndrome).
- Severe pulmonary dysfunction with little or no limb weakness at onset (think to myasthenia gravis).
- Normal nerve conduction tests after two weeks.
-
- Diagnosis
- Diagnosis is based on clinical evaluation and is then further confirmed and supported by CSF, electrodiagnostic *.
- The initial diagnosis of GBS is based upon clinical presentation (explained above).
- The clinical diagnosis is further supported if CSF shows albuminocytologic dissociation.
- Diagnosis is based on clinical evaluation and is then further confirmed and supported by CSF, electrodiagnostic *.
- Treatment
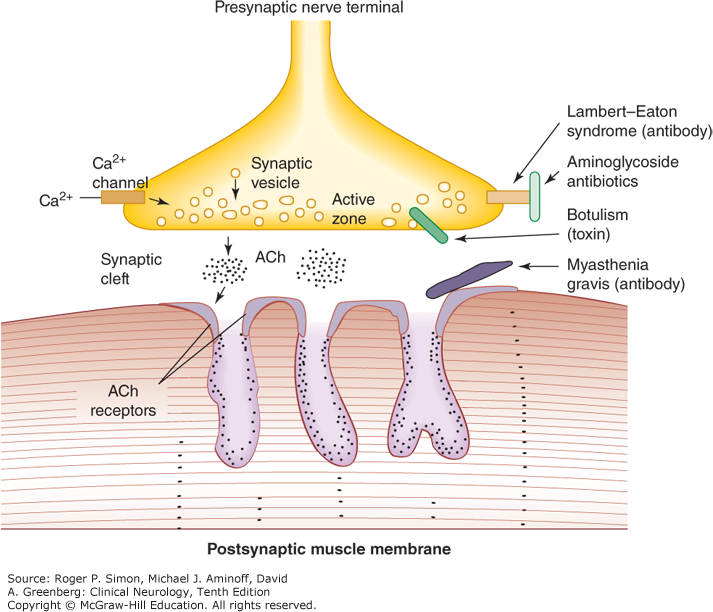
Myasthenia gravis
- Myasthenia gravis (MG) is the most common disorder of neuromuscular transmission.
- Pathophysiology
- Autoantibodies are formed which bind to acetylcholine receptors on skeletal muscle.
- Anti-acetylcholine antibodies impair transmission at the neuromuscular junction.
- Epidemiology: Can be associated with:
- Thymoma or thymic hyperplasia.
- Other autoimmune diseases (e.g., hyperthyroidism, lupus, rheumatoid arthritis, polymyositis).
- Lymphoma.
- Checkpoint inhibitor use
- Clinical presentation. There are two clinical forms of MG
- Ocular myasthenia
- Eyes and bulbar muscles tend to be involved early (ptosis and diplopia are common).
- Only skeletal muscles are involved (not the pupils).
- Isolated weakness in certain ocular muscles can occur, mimicking internuclear ophthalmoplegia or various cranial nerve palsies.
- Eyes and bulbar muscles tend to be involved early (ptosis and diplopia are common).
- Generalized disease
- Weakness commonly affects ocular muscles, but it also involves a variable combination of bulbar, limb, and respiratory muscles.
- Limb weakness is usually proximal > distal.
- Ocular myasthenia
- Typical features of weakness
- Fatigability: ongoing effort rapidly provokes worsening weakness. Strength improves with rest *
- Asymmetric and fluctuating in severity over time.
- Normal sensation and normal pupillary reflexes.
- Deep tendon reflexes are normal (until severe weakness occurs).
- DDx
- Drug-induced myasthenia, botulism, thyroid disorders, and other causes of ocular disorders, such as intracranial mass lesions.
- Cholinergic crisis
- Excessive doses of acetylcholinesterase inhibitor (e.g. pyridostigmine) lead to excessive levels of acetylcholine, which acts as a depolarizing paralytic (similar to succinylcholine).
- It can occur if patients are self-medicating with excessive doses of pyridostigmine, but doesn’t occur in patients taking standard pharmacologic doses of pyridostigmine (<120 mg every 3 hours).
- Clinical features *
- Fasciculation of skeletal muscles.
- Features of acetylcholine excess affecting the autonomic nervous system:
- Nausea/vomiting, diarrhea, salivation, lacrimation, diaphoresis.
- Miosis.
- Bradycardia.
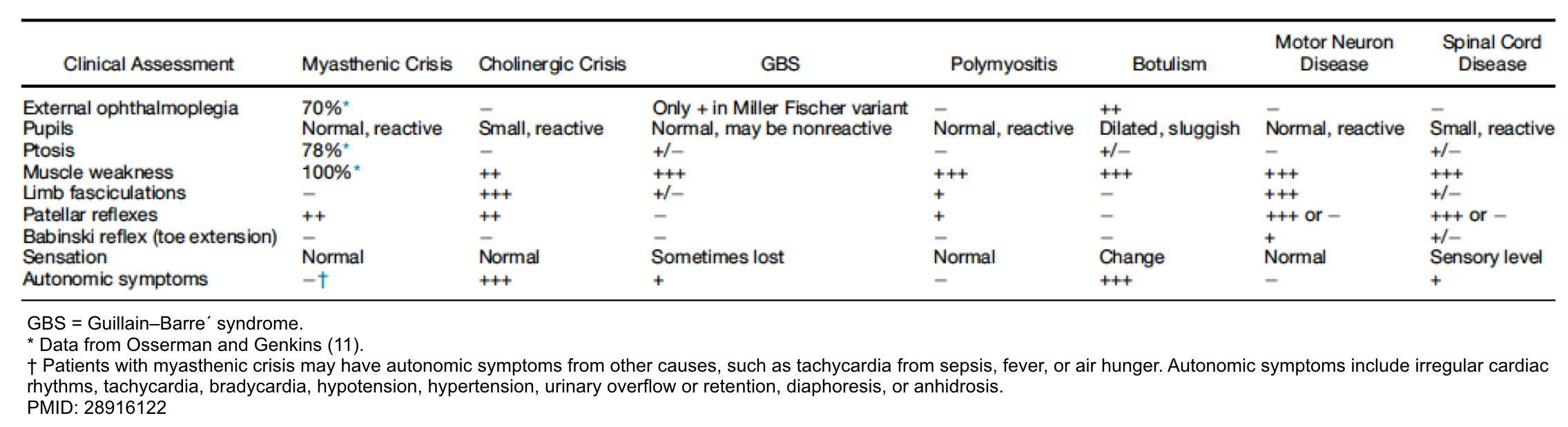
- Diagnosis
- Ice pack test
- Place ice pack over patient’s eye that is affected with ptosis or ophthalmoparesis for 2-5 minutes. Improvement following ice supports a diagnosis of myasthenia gravis (sensitive ~90% and specific ~80%) *.
- Electromyography: Repeated stimulation causes rapid deterioration in muscle responses (fatigability) *.
- Serologic testing for acetylcholine receptor antibodies.
- Ice pack test
- Myasthenic crisis
- Refers to extreme weakness in the muscles of respiration, resulting in respiratory failure.
- May happen due to exacerbation of disease in patients previously diagnosed with MG, or may be the initial manifestation of myasthenia gravis in about 15-20% of patients.
- Precipitating factors *
- Infections esp. pneumonia.
- Medications that exacerbate myasthenia.
- Nonadherence with myasthenia gravis medications (e.g. pyridostigmine).
- Tapering of immunosuppressive medications.
- Initiation or discontinuation of steroid.
- Electrolyte disturbances.
- Thyroid disease (either hypothyroid or hyperthyroid).
- Surgery/trauma/pregnancy.
- Diagnosis: requires two components:
- Other etiologies of cardiopulmonary disease (e.g. pneumonia, heart failure, pulmonary embolism) should be excluded. Therefore diagnostic evaluation include ECG, Bedside ultrasound, chest CT, and CT angiography to exclude PE (if clinically suspected).
- Evidence of worsening muscular weakness: History and physical may be helpful (e.g. patient reports increasing limb weakness and this is confirmed on examination).
- Differential diagnosis of myasthenic crisis
- Cholinergic crisis
- Thyroid storm or myxedema coma (remember that autoimmune thyroid disease is associated with myasthenia gravis).
- Adrenal crisis: Many patients with myasthenia are maintained on chronic prednisone for months or years. Adrenal crisis may occur in the following situations:
- Prednisone is abruptly stopped.
- Patient on chronic low-dose prednisone is exposed to a new source of physiologic stress.
- Management:
- Assess airway and breathing
- HFNC/BiPAP: For patients with “mild” respiratory distress; consider high-flow nasal cannula (HFNC) or BiPAP (if the patient is alert and oxygenating well).
- Intubation: decision to intubate should always be made on clinical grounds (i.e. it should not be made solely on the basis of pulmonary function tests). Worrisome signs of impending respiratory arrest include:
- Patient appearance: RR, worsening work of breathing
- Weak ability to cough
- Neck flexion weakness, which may tend to track with respiratory muscle weakness
- Progressively worsening hypoxemia: Generally, myasthenia shouldn’t cause significant hypoxemia. Progressively worsening hypoxemia is a worrisome sign that may suggest progressive atelectasis, aspiration, or an alternative overlooked diagnosis.
- Signs of worsening lobar collapse or aspiration on chest imaging.
- intubation procedure
- A non-depolarizing paralytic should be used (e.g. rocuronium) if ever.
- The dose should be reduced by ~50% compared to the usual dose (e.g. a dose of ~0.6 mg/kg rocuronium may be reasonable).
- Succinylcholine may fail to work due to reduced acetylcholine receptor density on muscle.
- Patients with dysautonomia due to GBS may be at risk of hypotension and/or bradycardia following intubation. Consider having epinephrine ready to manage this complication.
- A non-depolarizing paralytic should be used (e.g. rocuronium) if ever.
- Pyridostigmine
- New diagnosis of MG: May initiate at 60 mg PO q6hr *.
- Chronic MG and not intubated: Continue prior dose, unless it’s extremely high.
- May add glycopyrrolate to reduce oral secretions (e.g. 1 mg with each dose).
- Plasma exchange / IVIG: These help to produce rapid improvement, esp. in patients with myasthenic crisis *.
- Plasma exchange is preferred (it is the fastest approach to stabilize disease). A course of five exchanges (3–4 L per exchange) is generally administered over a 10- to 14-day period
- If plasma exchange is contraindicated/unavailable, may use IVIG. The usual dose is 2 g/kg, which is typically administered >2–5 days.
- Monitoring:
- Most useful: vital signs, clinical appearance, subjective dyspnea.
- Forced vital capacity 2-3 times daily.
- Assess airway and breathing
- Drugs to be avoid in MG. Many drugs can adversely affect neuromuscular function, leading to worsening of weakness and respiratory fatigue *.
1. Anticonvulsants: Phenytoin , ethosuximide, magnesium sulfate, barbiturates, lithium.
2. Antibiotics: Aminoglycosides, fluoroquinolones, macrolides.
3. Cardiovascular: Beta blockers, quinidine, lidocaine, procainamide, verapamil, statins.
4. Steroids: are standard treatment for myasthenia, but may cause transient worsening during the first two weeks.
5. Iodinated radiocontrast dye
6. Chloroquine and hydroxychloroquine.
7. Botulinum toxin
8. Desferrioxamine
9. D-penicillamine
10. Succinylcholine
11. Thyroid replacement therapy
12. Narcotic analgesics
13. Psychotropics: Haloperidol, lithium, amitriptyline.
13. Eye drops: Timolol, echothiophate
Botulism
- Botulism is a toxin-mediated neuromuscular junction disorder that causes acute weakness leading to respiratory failure.
- Botulinum toxin cleaves proteins required to release acetylcholine (ACh) from the cholinergic terminals within the motor end plate and within the peripheral parasympathetic nervous system (figure below). Regenerating this molecular machinery takes time, so acetylcholine transmission takes time to recover.
- Since botulinum toxin is a protein, it doesn’t penetrate the central nervous system.
- Epidemiology:
- Food botulism (preformed toxin in food, primary cause in adults):
- This is associated mostly with home-canned food (boiling without pressure-cooking kills most bacteria, but not C. botulinum spores). Other sources include fermented fish or seal meat, seal oil, and alcohol made from potatoes in prisons.
- Incubation period: Clinical botulism typically occurs ~12-36 hours after ingestion (although onset may be earlier, or delayed for up to ten days).
- Wound botulism
- This may result from subcutaneous injection of black tar heroin, or from severe trauma (e.g., extremity crush injury).
- Incubation period: Botulism may not occur for days or weeks after wound infection
- Iatrogenic botulism may result from use of excessive doses of commercial botulinum toxin (e.g., for cosmetic procedures).
- Food botulism (preformed toxin in food, primary cause in adults):
- Clinical presentation
- Descending symmetric flaccid paralysis:
- Paralysis almost always starts with cranial nerve involvement (e.g. ptosis, ophthalmoplegia, dysarthria, dysphagia, facial weakness) *.
- Subsequently, paralysis descends to involve the limbs.
- Reflexes are generally normal, unless there is severe weakness
- Peripheral anticholinergic autonomic effects:
- Dilated and nonreactive pupils (however, these are present in only half of patients).
- Pupillary involvement may be extremely helpful if present, since this is unlikely in many other conditions (e.g., myasthenia gravis) *.
- Dry mouth.
- Ileus, urinary retention.
- Dilated and nonreactive pupils (however, these are present in only half of patients).
- Food botulism: May additionally cause early gastrointestinal symptoms (nausea, vomiting, cramping, diarrhea)
- Wound botulism: Wound may look benign, or there may be associated features of fever and infection.
- Findings consistent with botulism *
- Normal mental status.
- Normal sensation (since the disorder is at the neuromuscular junction, there is no sensory deficit and no pain).
- Absence of fever (although fever can occur in wound botulism).
- Normal heart rate, or bradycardia.
- Normal blood pressure.
- Descending symmetric flaccid paralysis:
- Differential diagnosis: Other causes of descending weakness plus cranial nerve palsies:
- Organophosphate intoxication *.
- Presents with manifestations of cholinergic excess. The dominant clinical features include bradycardia, miosis, lacrimation, salivation, bronchorrhea, bronchospasm, urination, emesis, diarrhea, diaphoresis, and generalized weakness.
- Guillain-Barre syndrome (especially the Miller Fisher variant).
- Diphtheria infection.
- Myasthenia gravis (diplopia and ptosis are common).
- Tick paralysis.
- Afebrile ascending flaccid weakness, paralysis and ataxia, without sensory involvement.
- When the bulbar nerves and diaphragm become involved, respiratory failure ensues.
- Pontine infarction.
- Organophosphate intoxication *.
- Diagnosis: The diagnosis of botulism is made based on clinical findings and exclusion of other processes.
- Management
- Antitoxin: If clinical suspicion for botulism is high, antitoxin administration should not be delayed while awaiting additional diagnostic studies *.
- Wound botulism: Debridement plus antibiotic therapy (e.g., preferably penicillin G, or alternatively metronidazole).
- However, antibiotic should ideally be initiated after administration of antitoxin and wound debridement (since antibiotic administration may lyse bacteria, thereby releasing botulinum toxin).
- Food botulism
- There is no role for antibiotic therapy.
- Whole bowel irrigation may be considered to reduce ongoing absorption of botulinum toxin from the gut, if the contaminated food was recently ingested *.
- Secure the airway if there is respiratory compromise.
- Severe botulism may necessitate mechanical ventilation for months
Periodic paralysis
- Periodic paralysis refers a group of neuromuscular disorder diseases characterized by a defect in muscle ion channels.
- This can be categorized to
- Hypokalemic periodic paralysis
- Thyrotoxic periodic paralysis
- Hyperkalemic periodic paralysis
- Epidemiology:
- Most cases are hereditary (though some can be acquired e.g. thyrotoxic periodic paralysis).
- Clinical presentation
- Acute (attack) of generalized ascending flaccid paralysis involving proximal muscles > distal muscles *.
- Mild degree of bulbar and respiratory muscles involvement can occur infrequently.
- Level of consciousness and sensation are intact.
- Myotonia is a typical feature of hyperkalemic periodic paralysis.
- Precipitating factors are not very useful to distinguish the underlying class of periodic paralysis.
- All classes of periodic paralysis are provoked by period of rest after vigorous exercise and stress.
- High carbohydrate load may provoke hypokalemic periodic paralysis.
- Fasting and high potassium rich diet may provoke hyperkalemic periodic paralysis.
- Thyrotoxic periodic paralysis can be triggered by cold exposure, alcohol consumption, beta-2 agonist inhaler, pulse steroid and infection.
- Labs
- Low level of potassium is seen in hypokalemic and thyrotoxic periodic paralysis.
- Low level of potassium and elevated T4, T3 and low TSH are suggestive for thyrotoxic periodic paralysis.
- Diagnosis
- Hypokalemic periodic paralysis:
- When the familial hypokalemic periodic paralysis is diagnosed, acute classic attack of weakness does not require further evaluation. Otherwise the diagnosis of hypokalemic periodic paralysis requires documentation of low K during the attack.
- Keep in mind that between the attacks, serum K is normal in patients with hypokalemic periodic paralysis, while it serum K remains low in patients with secondary hypokalemia.
- Exercise test (running for 30min on treadmill) is a provocative test useful for diagnosis of hypokalemic periodic paralysis.
- Thyrotoxic periodic paralysis: Elevated T3,T4, decreased TSH and low level of serum K during the attack.
- Hyperkalemic periodic paralysis: Elevated serum K during the attack.
- Hypokalemic periodic paralysis:
- Management
- Hypokalemic periodic paralysis
- Document low serum K, before administration of potassium supplementation.
- KCl 10meq/h “PO” can be given to correct the serum potassium. Total dose of 60-120meq is usually required to abort the acute attack *. Cardiac monitoring is warranted during and after K supplementation.
- Thyrotoxic periodic paralysis
- Cardiac monitoring
- KCl 10meq/h “PO” maximum to total dose of 90meq in 24h can be given to correct the serum potassium. IV KCl may be required for severe hypokalemia and impaired swallowing.
- For refractory cases, propranolol 3mg/kg PO can be given *.
- Hyperkalemic periodic paralysis
- Hydrochlorothiazide 25-75mg PO
- Albuterol 0.1mg; 1-2 puffs *
- IV Calcium gluconate
- Hypokalemic periodic paralysis
Fascial nerve palsy
- Bell’s palsy is the most common cause of unilateral facial paralysis.
- Refer to (appendix 4) for function of fascial nerve and (appendix 5) for relevant anatomy of fascial nerve. The fascial nerve is a mixed nerve containing the following:
- Motor fibers that innervate the facial muscles
- Parasympathetic fibers innervating lacrimal, submandibular, and sublingual salivary glands
- Afferent fibers from taste receptors from the anterior two thirds of the tongue.
- Somatic afferents from the external auditory canal and pinna
- Etiology
- Idiopathic (aka Bell’s palsy)
- Infections:
- HSV reactivation, herpes zoster , HIV
- Lyme disease
- GBS
- Middle ear infection, mastoiditis
- Tumor: Mass lesions of temporal bone, internal acoustic canal, cerebellopontine angle, parotid gland; can compress or infiltrate the fascial nerve.
- Clinical features:
- Acute sudden onset of unilateral fascial paralysis, which may be preceded by pain around or behind the ear.
- Onset of facial paralysis is acute, with maximal symptoms in 2 to 3 days, and full or partial recovery within < 4-6 months.
- Fascial nerve paralysis may manifest as *
- Facial droop.
- Effacement of wrinkles and forehead burrows.
- Inability to completely close the eye.
- Facial numbness or hyperesthesia can accompany paralysis.
- Decreased tearing, hyperacusis, and/or loss of taste sensation on the anterior two thirds of the tongue may help to site the lesion in the fallopian canal, but these findings are of little practical use other than as indicators of severity.
- Subtle dysfunction of cranial nerves V, VIII, IX, and X may be associated.
- Diagnosis
- Diagnosis of idiopathic Bell’s palsy is based on the history and physical exam and is a diagnosis of exclusion of other conditions that can cause facial palsy.
- The diagnosis of Bell’s palsy is based upon the following criteria:
- There is a diffuse facial nerve involvement manifested by paralysis of the facial muscles, with or without loss of taste on the anterior two thirds of the tongue or altered secretion of the lacrimal and salivary glands.
- Onset is acute, over a day or two; the course is progressive, reaching maximal clinical weakness/paralysis within three weeks or less from the first day of visible weakness; and recovery or some degree of function is present within six months.
- A diagnosis of Bell’s palsy is doubtful if some facial function, however small, has not returned within 4 months.
- Differential diagnosis (table below)
- Middle cerebral artery ischemia or stroke
- Pontine stroke (where the facial nerve wraps around the abducens (cranial nerve VI) nucleus *.
- Peripheral facial nerve palsy and ipsilateral gaze palsy (inability to abduct an eye) due to ischemia of the cranial nerve VI nucleus.
- Contralateral hemiparesis, sensory loss, ataxia, nystagmus, ipsilateral abducens palsy are also present.
- GBS. Typically causes bilateral and symmetric fascial palsy accompanied by limb weakness, ophthalmoplegia and areflexia.
- Lyme
- Unilateral or bilateral fascial palsy can be preceded by painless nontender swelling and erythema of the face
- Other findings: Antecedent skin rash, arthritis, heart block vertigo and hearing loss.
- Tumor: Mass lesions of temporal bone, internal acoustic canal, cerebellopontine angle, parotid gland; can compress or infiltrate the fascial nerve.
- Clues:
- Prolonged, slowly progressive, relapsing or persistent paralysis without recovery.
- History of fascial twitch or spasm (suggesting ongoing fascial nerve irritation) preceding the fascial weakness .
- Hearing loss.
- Clues:
- Ear infection
- 👉Always perform an ear examination to identify otitis media and malignant otitis, and palpate the mastoids for tenderness, because infections of the ear and mastoids can affect the mastoid, tympanic, labyrinthine, or meatal segments of the cranial nerve VII.

- Indications for neuroimaging (contrast-enhanced CT or gadolinium-enhanced MRI):
- Atypical physical signs
- Any patient with facial paralysis sparing the forehead (CT or MRI to evaluate for stroke)
- Inability to abduct an eye (CT or MRI to evaluate for stroke).
- Presence of slow progression beyond 3 weeks
- Absence of improvement at 4 months.
- History of a facial twitch or spasm that precedes facial weakness
- Treatment
- Treatment with corticosteroids has shown significant benefit in the treatment of Bell’s palsy. Evidence from randomized controlled trials suggests a benefit when giving antiviral medications in combination with steroids over steroid therapy alone *.
- Prednisone 60-80mg/day for one week
- Valacyclovir 1000mg tds for one week; or
- Acyclovir 400mg five times daily for one week.
- Treatment with corticosteroids has shown significant benefit in the treatment of Bell’s palsy. Evidence from randomized controlled trials suggests a benefit when giving antiviral medications in combination with steroids over steroid therapy alone *.
-
- Patients with facial paralysis are at risk for corneal abrasions and keratitis due to eyelid weakness and incomplete eye closure.
- Patients can use a patch to close the affected eye if there is incomplete closure.
- Prescribe ocular lubricants, such as an artificial tear replacement, to prevent the eye from drying out.
- Patients with facial paralysis are at risk for corneal abrasions and keratitis due to eyelid weakness and incomplete eye closure.
Appendix
1. Myotome (the segmental innervation of skeletal muscle by ventral root of specific spinal nerve
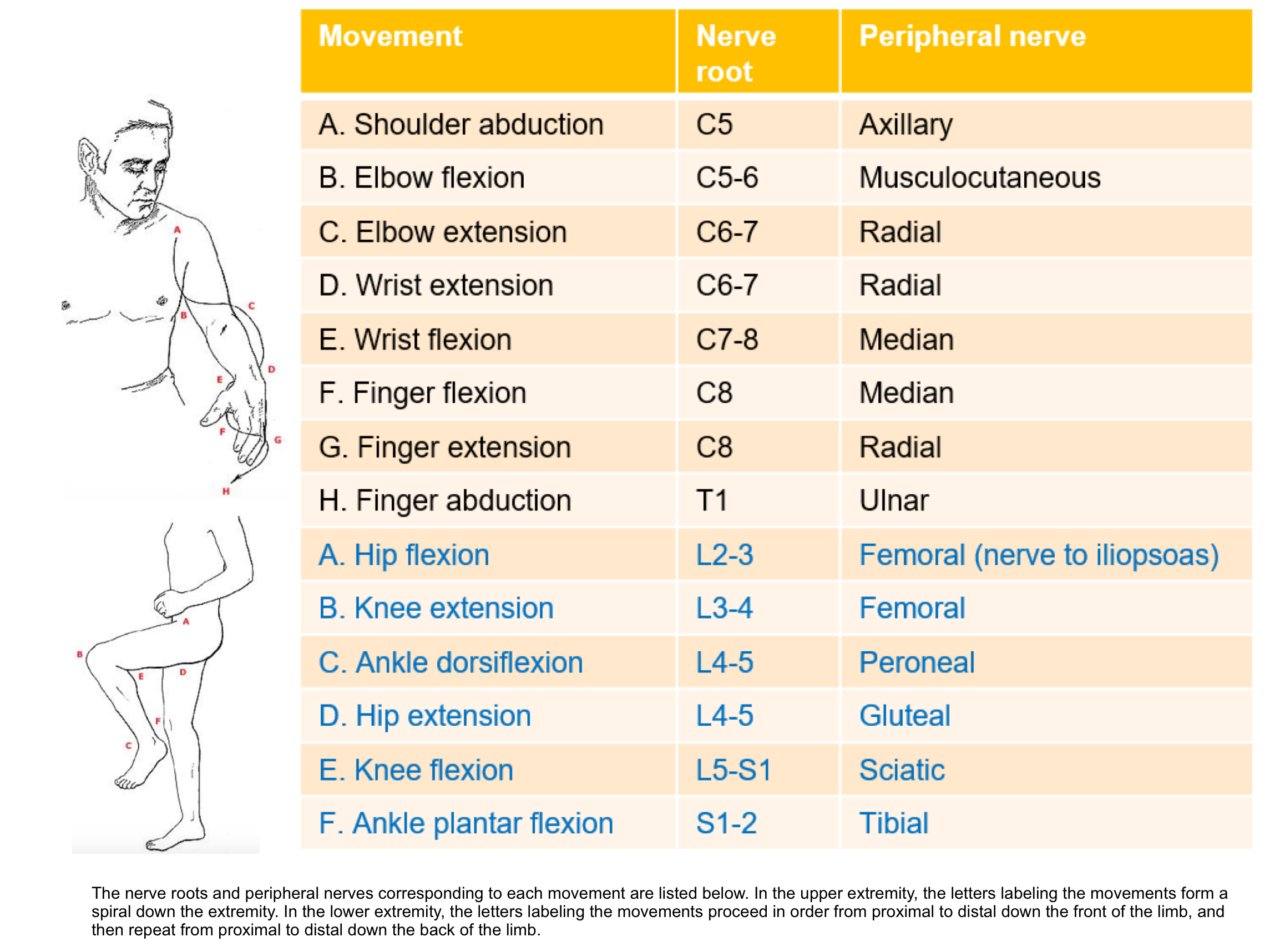
2. Peripheral neuropathy of upper and lower extremities

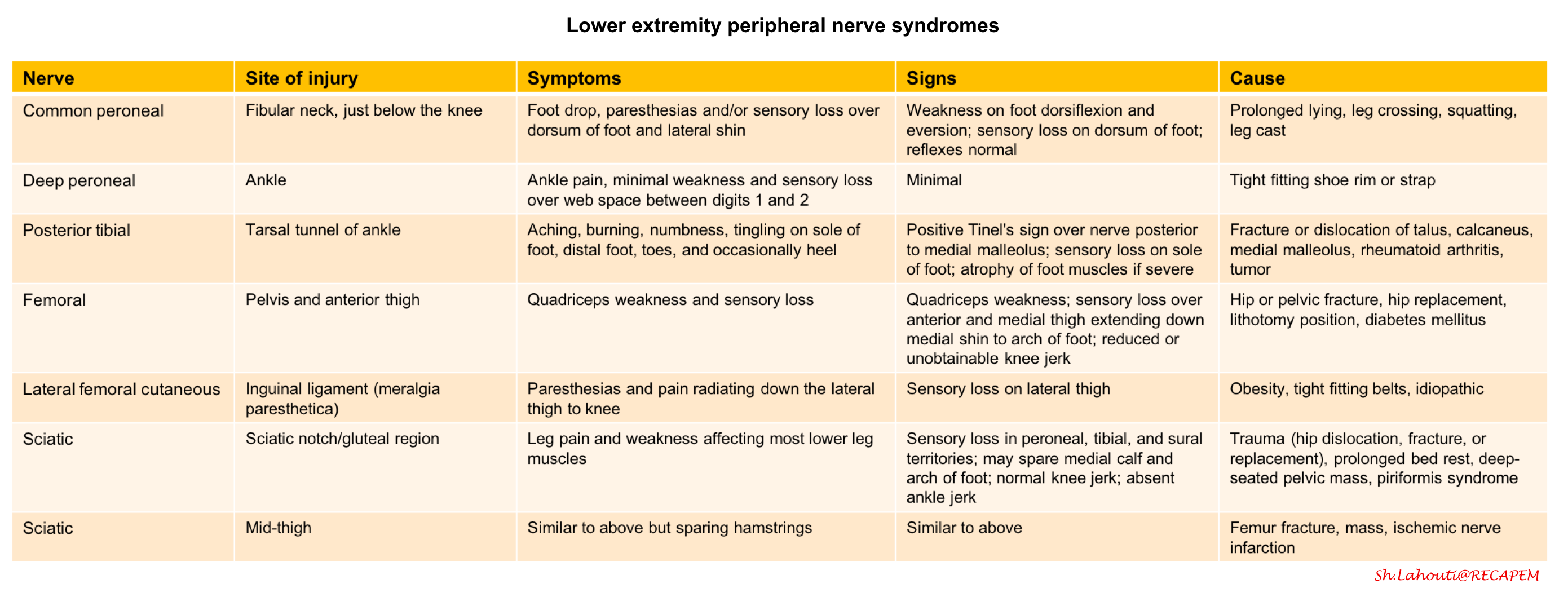
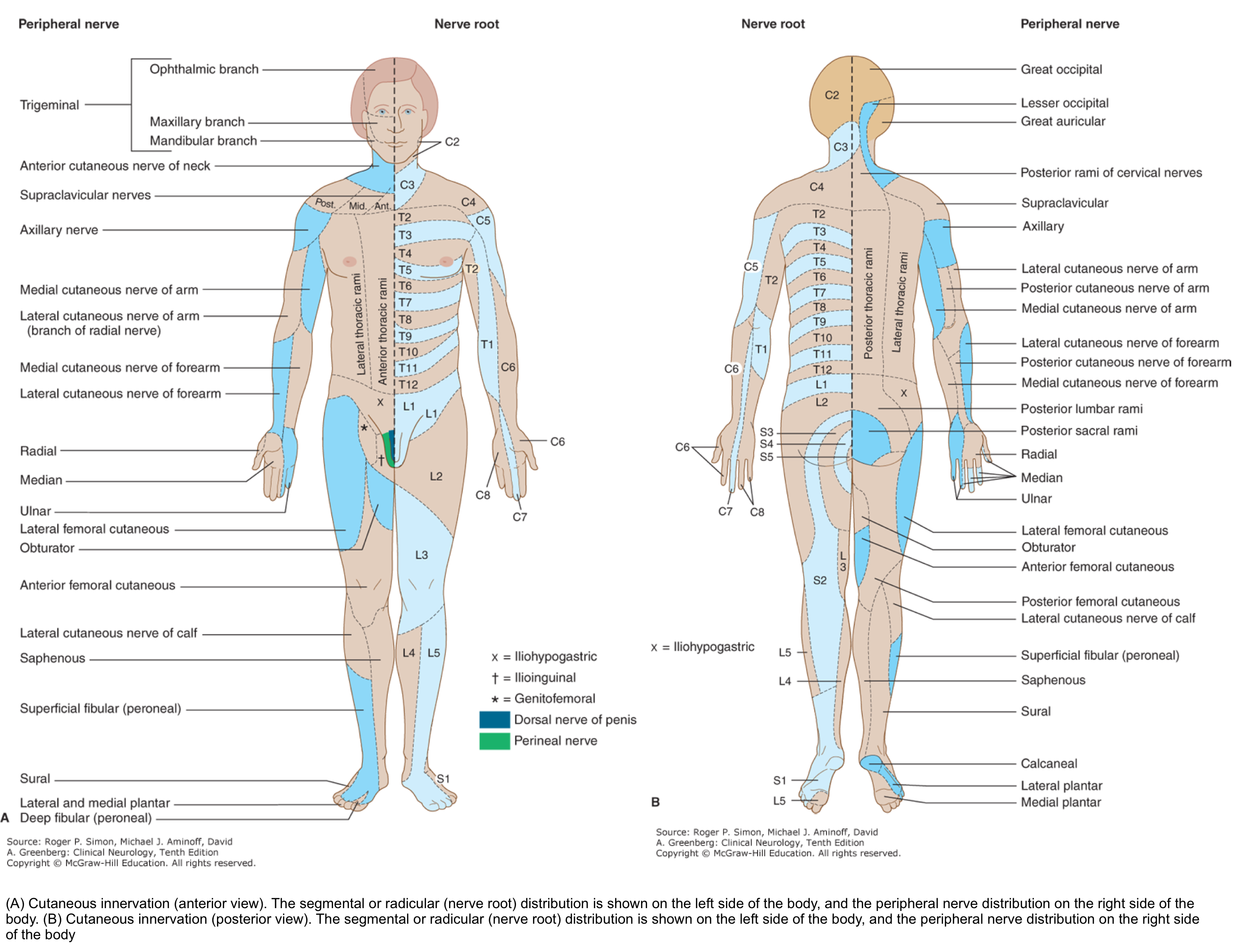
3.Dermatomes and peripheral nerve sensory innervation
3b. Localizing signs and symptoms of lumbosacral nerve root lesion

6. NMJ Disorder
Media
Brief Neurological examination
Babinski signs
Going further
- Approach to acute motor weakness (Emergency medicine cases)
- Neuromuscular Disorders & generalized weakness (IBCC)
References
1. Karakoumis J, Nickel CH, Kirsch M, Rohacek M, Geigy N, Müller B, Ackermann S, Bingisser R. Emergency Presentations With Nonspecific Complaints-the Burden of Morbidity and the Spectrum of Underlying Disease: Nonspecific Complaints and Underlying Disease. Medicine (Baltimore). 2015 Jul;94(26):e840. doi: 10.1097/MD.0000000000000840.
2. Sauter TC, Capaldo G, Hoffmann M, Birrenbach T, Hautz SC, Kämmer JE, Exadaktylos AK, Hautz WE. Non-specific complaints at emergency department presentation result in unclear diagnoses and lengthened hospitalization: a prospective observational study. Scand J Trauma Resusc Emerg Med. 2018 Jul 16;26(1):60. doi: 10.1186/s13049-018-0526-x.
3. LoVecchio F, Jacobson S. Approach to generalized weakness and peripheral neuromuscular disease. Emerg Med Clin North Am. 1997 Aug;15(3):605-23. doi: 10.1016/s0733-8627(05)70320-5.
4. Flower O, Bowles C, Wijdicks E, Weingart SD, Smith WS. Emergency neurological life support: acute non-traumatic weakness. Neurocrit Care. 2012 Sep;17 Suppl 1:S79-95. doi: 10.1007/s12028-012-9752-7.
5. Peeling JL, Muzio MR. Conversion Disorder. 2022 May 15. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan.
6. Mehndiratta MM, Kumar M, Nayak R, Garg H, Pandey S. Hoover’s sign: Clinical relevance in Neurology. J Postgrad Med. 2014 Jul-Sep;60(3):297-9. doi: 10.4103/0022-3859.138769.
7. Singh TD, Wijdicks EFM. Neuromuscular Respiratory Failure. Neurol Clin. 2021 May;39(2):333-353. doi: 10.1016/j.ncl.2021.01.010.
8. Sheikh KA. Guillain-Barré Syndrome. Continuum (Minneap Minn). 2020 Oct;26(5):1184-1204. doi: 10.1212/CON.0000000000000929.
9. Khamees D, Meurer W. Approach to Acute Weakness. Emerg Med Clin North Am. 2021 Feb;39(1):173-180. doi: 10.1016/j.emc.2020.09.010. Epub 2020 Nov 5.
10. Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-Barré syndrome. Lancet. 2021 Mar 27;397(10280):1214-1228. doi: 10.1016/S0140-6736(21)00517-1. Epub 2021 Feb 26.
11. Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barré syndrome. Lancet. 2016 Aug 13;388(10045):717-27. doi: 10.1016/S0140-6736(16)00339-1. Epub 2016 Mar 2.
12. Walters J. Weakness in the intensive care unit. Pract Neurol. 2022 Jul 21:pn-2022-003422. doi: 10.1136/pn-2022-003422.
13. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2014 Sep 19;2014(9):CD002063. doi: 10.1002/14651858.CD002063.pub6.
14. Hughes RA, Brassington R, Gunn AA, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2016 Oct 24;10(10):CD001446. doi: 10.1002/14651858.CD001446.pub5.
15. Roper J, Fleming ME, Long B, Koyfman A. Myasthenia Gravis and Crisis: Evaluation and Management in the Emergency Department. J Emerg Med. 2017 Dec;53(6):843-853. doi: 10.1016/j.jemermed.2017.06.009. Epub 2017 Sep 12.
16. Ciafaloni E. Myasthenia Gravis and Congenital Myasthenic Syndromes. Continuum (Minneap Minn). 2019 Dec;25(6):1767-1784. doi: 10.1212/CON.0000000000000800.
17. Witoonpanich R, Dejthevaporn C, Sriphrapradang A, Pulkes T. Electrophysiological and immunological study in myasthenia gravis: diagnostic sensitivity and correlation. Clin Neurophysiol. 2011 Sep;122(9):1873-7. doi: 10.1016/j.clinph.2011.02.026. Epub 2011 Mar 17.
18. Wendell LC, Levine JM. Myasthenic crisis. Neurohospitalist. 2011 Jan;1(1):16-22. doi: 10.1177/1941875210382918.
19. Sheikh S, Alvi U, Soliven B, Rezania K. Drugs That Induce or Cause Deterioration of Myasthenia Gravis: An Update. J Clin Med. 2021 Apr 6;10(7):1537. doi: 10.3390/jcm10071537.
20. Guidon AC. Lambert-Eaton Myasthenic Syndrome, Botulism, and Immune Checkpoint Inhibitor-Related Myasthenia Gravis. Continuum (Minneap Minn). 2019 Dec;25(6):1785-1806. doi: 10.1212/CON.0000000000000807.
21. Guidon AC. Lambert-Eaton Myasthenic Syndrome, Botulism, and Immune Checkpoint Inhibitor-Related Myasthenia Gravis. Continuum (Minneap Minn). 2019 Dec;25(6):1785-1806. doi: 10.1212/CON.0000000000000807.
22. Zanin A, Sartori S, Salandin M, Laverda AM, Fenicia L, Anniballi F, Cogo PE. A descending cranial nerve palsy during the christmas holidays. Neurohospitalist. 2012 Apr;2(2):66-70. doi: 10.1177/1941874412438903.
23. Edmundson C, Bird SJ. Acute Manifestations of Neuromuscular Disease. Semin Neurol. 2019 Feb;39(1):115-124. doi: 10.1055/s-0038-1676838. Epub 2019 Feb 11.
24. Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006 Jan;129(Pt 1):8-17. doi: 10.1093/brain/awh639. Epub 2005 Sep 29.
25. Levitt JO. Practical aspects in the management of hypokalemic periodic paralysis. J Transl Med. 2008 Apr 21;6:18. doi: 10.1186/1479-5876-6-18. Erratum in: J Transl Med. 2014;12:198. Dosage error in article text.
26. Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia, and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001 Mar;37(3):620-3.
27. Gilden DH. Clinical practice. Bell’s Palsy. N Engl J Med. 2004 Sep 23;351(13):1323-31. doi: 10.1056/NEJMcp041120.
28. Gronseth GS, Paduga R; American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012 Nov 27;79(22):2209-13. doi: 10.1212/WNL.0b013e318275978c. Epub 2012 Nov 7.
Add comment